Course Authors
Ogo I. Egbuna, M.D., M.Sc., and Edward M. Brown, M.D.
Release Date: 07/15/2008
Upon completion of this Cyberounds®, you should be able to:
Describe Ca2+o homeostasis, calcium-sensing receptors (CaSRs) and the role of the prototypical CaSR in Ca2+o homeostasis
Discuss recent advances in our understanding of the structure-function relationships and ligand binding sites of the CaSR
List the spectrum of mutations involving the CaSR that cause human disease and describe disorders arising from anti-CaSR autoantibodies
Discuss the actions of the CaSR in CaSR-expressing tissues in defending against both hypo- and hypercalcemia
Discuss the current and potential utility of CaSR-based therapeutics
Outline future directions in CaSR physiology and research.
The calcium-sensing receptor (CaSR) plays key roles in the maintenance of a narrow range (1.1-1.3 mM) of extracellular ionized calcium concentration (Ca2+o), primarily by modulating the function of chief cells of the parathyroid gland. The CaSR regulates the synthesis and secretion of parathyroid hormone (PTH), as well as parathyroid cellular proliferation, inhibiting all three processes when Ca2+o is high and stimulating them when Ca2+o is low.(1) It serves as a "calciostat," informing the parathyroid glands and other tissues where it is expressed of the precise level of Ca2+o.
The CaSR (also known as CaSR1 or GPRC2A) was identified using the expression-cloning technique in Xenopus laevis (the African clawed frog) oocytes.(2) It is a member of family C of the superfamily of 7-transmembrane, G protein-coupled receptors (GPCRs). Other members of this family are the so-called metabotropic receptors for glutamate (mGluRs), receptors for gamma-aminobutyric acid (GABA), as well as GPCRs for sensing pheromones, taste and odorants (in fish). Recently, another member of family C, GPRC6A, has been found to share several pharmacological properties with the CaSR.(3),(4) Like the CaSR, GPRC6A is sensitive towards certain L-amino acids but is most responsive to basic amino acids and less so to calcium;(3),(5) nevertheless, it is possible that GPRC6A is a second calcium-sensing receptor (CaSR2).
The physiological relevance of the CaSR in humans has been proven by experiments-of-nature in inherited human disorders caused by mutations in the receptor leading to either loss- or gain-of-function.(6) Heterozygous inactivating mutations of the CaSR cause a disorder of calcium metabolism, familial hypocalciuric hypercalcemia (FHH), that manifests as asymptomatic hypercalcemia with relative or absolute hypocalciuria. Homozygous mutations, on the other hand, cause a severe, sometimes lethal form of hyperparathyroidism, with marked hypercalcemia and hyperparathyroidism if left untreated. Activating mutations, in contrast, cause an autosomal dominant form of hypocalcemia (ADH) associated with low normal or low PTH levels, despite hypercalcemia and relative or absolute hypercalciuria.
CaSR expression is greatest in the parathyroid glands, calcitonin-secreting C-cells of the thyroid gland and kidney. The CaSR is also found in the two other key organs that participate in calcium homeostasis: gut and bone.(7) Available data have demonstrated that the CaSR is expressed not only in the organs that secrete calcium-regulating hormones (e.g., the parathyroid glands and C-cells of the thyroid glands) but also in target tissues for these hormones and in tissues not known to be involved in Ca2+o homeostasis (see Figure 1).(8) These latter tissues regulate Ca2+o by translocating calcium ions into or out of the bodily fluids and include the kidney, which expresses the CaSR at robust levels in certain nephron segments, as well as bone and intestine, which express the receptor at lower levels. By acting on both hormone-secreting and hormone-responsive, calcium-transporting tissues through its own cell surface receptor, Ca2+o acts, in effect, as another Ca2+o-regulating "hormone" (in this case Ca2+o-lowering) or "first messenger."

Figure 1. Tissue Distribution and Density of Expression of the CaSR.
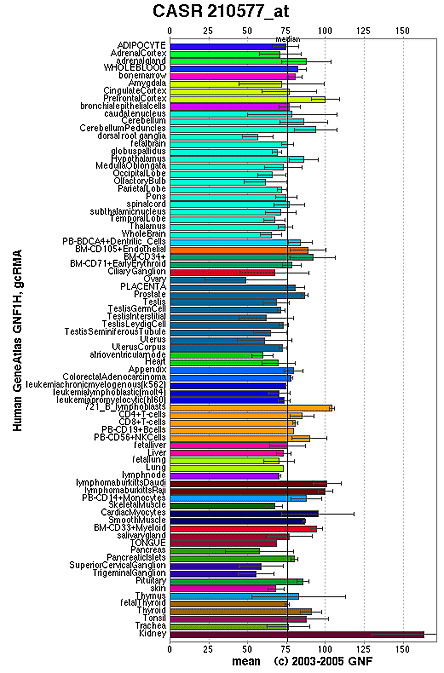
Adopted with permission from the Novartis Research Foundation Gene Atlas Database.(8)
Current Understanding of Ligand Binding Sites of the CaSR
The human CaSR has a large extracellular domain (ECD) (612 residues) -- a transmembrane domain (TMD) of 250 amino acids containing the 7-membrane spanning helices characteristic of the GPCRs and an intracellular C-terminal domain (ICD) of 216 amino acids. The receptor exhibits substantial N-linked glycosylation, which is important for normal cell membrane expression of the receptor but does not appear to modify the function of the receptor per se.(9) The functional cell surface form of the CaSR is a dimer and the two monomers within the dimeric CaSR are linked by disulfide bonds involving cysteine residues 129 and 131 within each monomer (see Figure 2A).(10) The ECD of each CaSR monomer consists of a bilobed, venus fly trap motif (VFTM), with a crevice between the lobes likely participating in ligand binding, and a cysteine-rich region just proximal to the first transmembrane domain.
Extracellular calcium is the prototypical ligand that activates the CaSR, although several other polyvalent cations and polycationic amine ligands have been identified that activate, inactivate or allosterically modulate the receptor.(7) The ECD of the CaSR likely contains more than one binding site for Ca2+o because the Hill coefficient (a measure of cooperativity in binding of a ligand to its receptor) for the activation of the receptor by Ca2+o is 3-4, consistent with the presence of positive cooperativity among at least this number of binding sites within the dimeric CaSR.(11),(12) The TMD is also apparently involved in Ca2+o-sensing, since a mutant CaSR lacking the ECD also responds to Ca2+o and other polyvalent cations.(13)
A major barrier to advancing our understanding of the role of Ca2+o in the regulation of the CaSR is our incomplete understanding of its Ca2+o binding sites, which is largely hindered by the lack of a solved three-dimensional structure and rapid association-dissociation binding rates for Ca2+o as a result of low binding affinities. Delineation of the Ca2+ binding pocket within the conserved VFTM of the human CaSR has been attempted using molecular modeling, as well as mutational and functional analyses, and is thought to reside in a crevice between lobes 1 and 2 of the VFTM(14) that appears to be conserved within several other class III GPCRs that are also responsive to Ca2+o. This information provides the foundation for an eventual molecular understanding of the effects of Ca2+ on these receptors.
Further insight into the three predicted binding sites [located in lobes 1 and 2 and in the crevice between the two lobes (Figure 2B-C)] has been derived from recent studies utilizing site-directed mutagenesis, nuclear magnetic resonance spectroscopy, fluorescent energy transfer analysis and insertion of peptides containing putative Ca2+o-binding sites into a scaffold protein to identify Ca2+-binding sites in the ECD of the CaSR.(15) The importance of these sites for binding to Ca2+ was confirmed by directed mutagenesis of amino acids present in these sites and the observation of significant potentiating and inhibiting effects on the EC50 (half maximal effective concentration) for Ca2+o-induced alterations in the function of the receptor. For further insights, the reader is referred to reviews of structure-function relationships of the CaSR from mutations and allosteric modifiers, as well as clinical conditions resulting from mutations of the CaSR.(16),(17)
Figure 2. Structure of the CaSR Venus Flytrap Homodimer
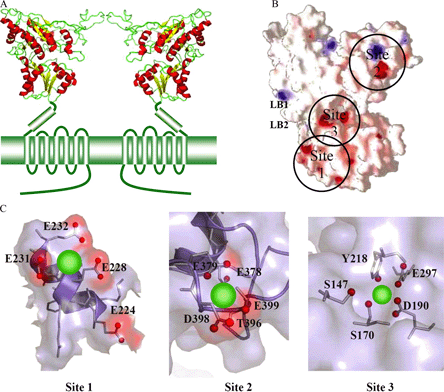
Printed with permission of the authors.(15)
Printed with permission of the authors.(15)
The Genetics and Spectrum of Mutations of the CaSR in Human Disease
Two hundred and thirteen mutations have been described for the CaSR (188 missense, 17 nonsense, six insertion and/or deletion, one silent and one splice mutation), which can be found in the CaSR mutation database (www.casrdb.mcgill.ca). These mutations are the molecular basis for inherited diseases of Ca2+o-sensing found in families with FHH (familial hypocalciuric hypercalcemia), NSHPT (neonatal severe hyperparathyroidism) and ADH or as comparable conditions arising from de novo mutations.(18) The majority of these mutations are located in the ECD, with the TMD representing the second most common site of mutations.
Most of the mutations found in the ECD are located in the first third of the N-terminus, suggesting the importance of this region in ligand binding. Activating mutations in the proximal one-third of the ECD may facilitate the ligand binding interaction in the different binding sites, increasing the receptor's affinity for its ligand, perhaps by stabilizing the active conformation of the receptor. Conversely, inactivating mutations may have the opposite effect, disrupting ligand binding pockets or stabilizing the inactive conformation of the receptor. This is supported by in vitro functional analyses of mutations in this location that show ligand-dependent changes in the apparent affinity of the receptor for Ca2+o.(16),(19) In addition to these mechanisms for receptor inactivation, some mutations produce receptors that never reach the cell surface or totally lack the TMD (e.g., due to truncation mutations), or have sufficient disruption of their structure such that they totally lose the capacity to bind Ca2+o or are unstable and are degraded during their biosynthesis.
Activating mutations in the TMD may negate constraints on the conformation of the CaSR that would otherwise maintain it in the inactive conformation that is present with low levels of Ca2+o. For example, residues in TM7 are critical for maintaining the receptor in an inactive conformation.(13) From functional analyses of receptors with gain-of-function mutations, the mutation A843E showed the ability to activate the receptor in the absence of the ligand, providing the only example to date of a naturally occurring mutation producing constitutive (constant) activation.(18)
The other activating mutations all showed ligand-dependent shifts of their dose-response curve to the left. Except for the A843E mutation, which apparently locks the TMD in the active conformation, these observations suggest that the mechanism of activation of the receptor in most of the TMD mutations is to increase apparent affinity and/or facilitate conformational changes in the TMD necessary for activation. Inactivating mutations in the TMD, in contrast, may prevent these same conformational changes, even though the receptor is well expressed in the plasma membrane.(20)
The ICD seems to be important for receptor trafficking to the cell membrane and for the interaction with G proteins and the CaSR's other intracellular binding proteins. A large deletion of the c-terminal tail was associated with gain-of-function in an ADH family.(21) In vitro functional studies showed gain-of-function and increased mutant receptor cell surface expression level, which may have contributed to the left shift in EC50, separate from any accompanying change in the intrinsic affinity of the mutant receptor. Mutagenesis in the ICD confirms its involvement in degradation and processing of the receptor.(21) Residues 962-981 in the c-terminal tail are critical for its interaction with filamin A, and this interaction markedly retards the receptor's degradation and facilitates MAPK signaling.(22) In contrast, interaction of the ICD with dorfin, an E3 ubiquitin ligase, targets the receptor for degradation.(23)
The persistence of the CaSR on the plasma membrane of the parathyroid cell may be key for its function as "calciostat". This could result from ligand-induced internalization of the receptor, followed by its recycling to the cell surface at an equivalent rate, as well as simply by a prolonged residence times of the CaSR on the plasma membrane.
Activating Mutations
Several (over three dozen) activating mutations in the CaSR have been characterized. The majority are missense mutations, with two deletions described. Most ADH-affected individuals are heterozygous for the activating mutation. In one family, homozygous mutation was described in one family member but it was not associated with a more severe phenotype.(21) Clinical data from affected individuals with activating mutations are abundant. In general, the more severe the hypocalcemia, the more likely patients are to have classical symptoms and complications of hypocalcemia, including tetany, seizures and basal ganglia calcification. ADH patients are particularly prone to develop renal complications during treatment with vitamin D and calcium supplementation (i.e., nephrocalcinosis and nephrolithiasis) and should only be treated if symptomatic and only to the point where symptoms disappear, with close monitoring of serum and urine calcium.
Inactivating Mutations
Numerous inactivating mutations of the CaSR have also been described. Fifty-nine are missense mutations, six are nonsense mutations, six are insertions and/or deletion including an Alu element insertion(24) and one is a splice mutation.(25) A number of dominant negative mutations, S137P, R185Q, R227L, R795W and F881L, have been described and can produce levels of hypercalcemia in families with these mutations higher than the norm. Even in the latter cases, however, the condition is generally asymptomatic with a benign clinical course and patients should simply be followed expectantly without parathyroid surgery.
There is a clear gene dosage effect with inactivating mutations of the CaSR: heterozygous inactivating mutations leading to FHH, with mild hypercalcemia, while homozygous mutations result in neonatal severe hyperparathyroidism (NSHPT), a more severe phenotype that manifests very early in life with severe hypercalcemia, bone demineralization and failure to thrive.(26) In such cases, if they do not respond to vigorous hydration, a bisphosphonate and/or perhaps a calcimimetic (see below), then total parathyroidectomy, with or without autotransplantation of a portion of the excised parathyroid tissue, may be essential for the newborn's survival.
However, three cases of de novo NSHPT reported in the literature are heterozygous for missense mutations located in the ECD, with only one mutated allele and no mutation found in the parents.(27),(28) One of these cases was heterozygous for a previously described mutation in another, apparently unrelated FHH family.(27) Because these heterozygous patients generally have a less severe form of hyperparathyroidism than the homozygous form of NSHPT, their condition is now often called neonatal hyperparathyroidism (NHPT). In fact, if treated with aggressive medical therapy, they may revert to a clinical picture of FHH over time.
Polymorphisms Involving the CaSR
Six single nucleotide polymorphisms (SNPs) have been found in the CaSR gene: one in intron 5 just before exon 6 (IVS 5 -88 t/c) and the remaining five in exon 7 in the coding region (one in the 6th TM [A826T -- given as amino acid change], one in the 7th TM [C851S] and three in the ICD [A986S, R990G and Q1011E]). The polymorphism in intron 5, IVS 5 -88 t/c is very common(29) but no correlation has been found between this mutation and the incidence of parathyroid adenoma or diabetes.(30) The A826T mutation has been seen in 16% of 50 normal subjects.(31) The C851S mutation was found in an ADH family in both affected and unaffected members(32) but the investigators also found another mutation in this family (A116T), which segregated with the disease, and the authors concluded that C851S was a rare polymorphism. The frequency of three common polymorphisms in the cytoplasmic tail varies in different populations. In one investigation of 377 unrelated DNA samples in a normal Caucasian (Italian) population, the relative frequencies for the CaSR SNPs producing the 986S, 990G and 1011E minor alleles were 24%, 4% and 3% respectively.(33)
At the present time, there is little evidence that the SNPs directly cause disease but there is the possibility that they may be associated with increased risk for disease. For example, on the basis of a CaSR polymorphism haplotype study in stone-forming patients, it was suggested that the 990G variant could influence renal CaSR activation and calcium excretion.(34) There is increased severity of primary hyperparathyroidism in patients who are G/G at codon 990(35) and patients with the A/A genotype at codon 986 or Q/Q genotype at codon 1011 have slightly lower serum calcium concentrations, albeit within the normal range, than the other genotypes.(33) For additional information, the reader is referred to an excellent review by Yun et al.(36)
Parathyroid and CaSR Antibodies: Growing Insights
There has been increasing clarity in the understanding of immunological perturbations involving the parathyroid gland and calcium homeostasis but controversy remains over the place of antibodies as the cause or result of parathyroid disease. Patients with endocrinopathies linked to autoimmune polyglandular syndrome (APS) often have a higher prevalence of organ-specific autoantibodies with specificities to autoantigens that are different from that observed in patients with isolated endocrinopathies without the APS. Recent important observations in this regard have involved the identification of non-cytotoxic antibodies that are specific for the CaSR and exert functional actions on the parathyroid(37),(38),(39) as opposed to destructive cytotoxic antibodies directed to autoantigens in the parathyroid cell.(40),(41)
As early as the 1940s, autoimmune destructive parathyroid disease was recognized as evidenced by fatty infiltration, lymphocytic infiltration and atrophy.(42),(43),(44) The first description of autoantibodies involving the parathyroid gland date back to 1966 when Blizzard and colleagues, using indirect immunoflourescence techniques, identified autoantibodies to the parathyroid gland in 33% of patients with idiopathic hypoparathyroidism.(45) Several years later, however, Swana(46) and later Betterle,(47) using similar methods, identified anti-mitochondrial antibodies to the oxyphil cells of the parathyroid. They suggested that these antibodies could explain the seropositivity seen by Blizzard but were not specific for the parathyroid per se.
In 1996, soon after the cloning of the CaSR, Li and colleagues identified the CaSR as an autoantigen in over half of their patients with autoimmune hypoparathyroidism.(48) In retrospect, a study carried out almost a decade earlier described autoantibodies recognizing 200kDa and 130kDa autoantigens in the parathyroid glands of patients with idiopathic hypoparathyroidism. These autoantibodies may have been interacting with the dimeric and monomeric forms of the CaSR, which have similar molecular weights.(49) CaSR-specific antibodies which activate the receptor have subsequently been shown to increase inositol phosphate (through activation of phospholipase C), activate the extracellular signal-regulated kinases 1 and 2 and inhibit PTH secretion, consistent with the ability of these antibodies, in contrast to cytotoxic antibodies, to alter parathyroid cell function while maintaining cell viability.(37) Inactivating antibodies, producing PTH-dependent hypercalcemia, have exactly the opposite effects.
Of equal interest are the observations of associations of CaSR-specific seropositivity in patients with sporadic idiopathic hypoparathyroidism (SIH) with major histocompatibility complex human leucocyte antigen (HLA) class II DR loci. The HLA-DR loci have been associated with several well known autoimmune diseases such as systemic lupus, psoriasis and rheumatoid arthritis. The frequency of HLA-DRB1 x 09 or DRB1 x 10 was found to be higher in CaSR seropositive patients with an odds ratio of 5.2 which, however, did not segregate patients for well known clinical manifestations of sporadic idiopathic hypoparathyroidism.(38) Like other autoimmune diseases, sporadic idiopathic hypoparathyroidism secondary to CaSR-specific antibodies is likely polygenic in nature with a probable role for environmental risk factors in the clinical manifestation of the disease, as one patient with these antibodies is reported to have had a spontaneous remission of disease.(37) Finally, in studies by Blizzard(45) and Goswami,(38) 6% and 13% of healthy controls were positive for parathyroid or CaSR autoantibodies, respectively, raising the hypotheses of the need for other environmental or genetic triggers for full expression of the phenotype of disease related to the presence of these antibodies.
Actions of the CaSR in Defending Against Both Hypo- and Hypercalcemia
In our studies characterizing mice with homozygous knock-out of the CaSR and/or the PTH genes, we observed altered homeostatic responses to exogenous 1,25(OH)2D3, PTH administration or high calcium diets as a result of the absence of one or both genes.(50),(51),(52) We used three genotypes of mice [(CaR+/+PTH-/- called C+P-), (CaR-/-PTH-/- called C-P-) or wild type mice (CaR+/+PTH+/+ called C+P+)] to evaluate these responses in vivo and made comparisons between:
- the C+P- and C-P- mice, in order to assess the role of CaSR independent of the effects of PTH;
- C+P+ mice and C+P- mice, to assess the role of CaSR-regulated PTH secretion.
Mice with homozygous knockout of the CaSR gene (CaR-/-PTH+/+, or C-P+) are profoundly hypercalcemic from uncontrolled hyperparathyroidism and die within the first few days to weeks of birth and as such have not been viable tools for investigation in our studies.
On a normal calcium diet and plain water, the C+P- and C-P- mice were both hypocalcemic as a result of the coexistent hypoparathyroidism (~6 mg/dl) compared to C+P+ mice who were normocalcemic. Both C+P- and C-P- mice became more hypocalcemic (~5 mg/dl) when placed on a calcium-deficient diet and plain water compared to C+P+ mice (~7.8 mg/dl), who have maximal PTH secretion and renal retention of calcium, as well as increased bone resorption. Challenge of these mice with an oral calcium load in the drinking water (1% and 2% CaCl2 consecutively) revealed a markedly blunted calcitonin response in the C-P- mice but normal responses (3-10 fold increase) in the C+P- and C+P+ mice. The responses to the oral calcium load also included a significant right shift in the relationship between serum calcium and the urinary calcium-to-creatinine ratio in the C-P- relative to the C+P+ and C+P- mice. Furthermore, there were virtually no increases in urinary calcium excretion with profound increases in serum calcium in the PTH-infused C-P- mice.
After intraperitoneal injection of a supraphysiologic dose of 1,25(OH) 2D3, peak calcemic responses were observed at 24 hours post injection with:
- no significant increase in serum calcitonin secretion in the C+P+ and C+P- mice, in contrast to the change observed upon exposure to a high oral calcium load;
- a nearly 2-fold greater increase in serum calcium levels in the C-P- mice compared to other genotypes on a calcium replete diet that was attenuated by use of a calcium-deficient diet and anti-resorptive agents; and
- increased urinary calcium to creatinine ratios in the C+P- and C+P+ mice but a decrease in C-P- mice on a Ca replete diet.
These studies strongly suggest that CaSR-stimulated PTH release provides a "floor" that defends against hypocalcemic challenges, which is absent in the hypoparathyroid C+P- and C-P- mice. In contrast, even in the absence of PTH, the CaSR provides a "ceiling," limiting the rise in serum calcium in response to a calcium load, comprising CaSR-stimulated calcitonin secretion, CaSR-induced renal calcium excretion and probably CaSR-mediated blunting of vitamin D3 action. This model of the CaSR -- playing distinct roles in providing a "ceiling" and "floor" for maintaining serum calcium homeostasis -- differs from traditional models in which the receptor provides continuous responses of Ca2+o-elevating and -lowering hormones and other Ca2+o-regulatory mechanisms to changes in Ca2+o, which all contribute to the homeostatic responses to both hypo- or hypercalcemia.
CaSR-based Therapeutics
Within the past several years, CaSR-based therapeutics have moved into the clinical arena with the approval by the FDA of the calcimimetic, cinacalcet (Sensipar® in the U.S. and Mimpara® in Europe)(53) for the treatment of uremic secondary hyperparathyroidism in patients receiving dialysis treatment (e.g., with stage 5 chronic kidney disease[CKD]),(54) as well as in parathyroid cancer. Calcimimetics are allosteric activators of the CaSR that bind to a site on the receptor within its TMD, which is distinct from that/those binding calcium. In stage 5 CKD, the calcimimetic lowers serum PTH, serum calcium, phosphorus and the calcium-phosphorus product, enabling a greater percentage of patients to achieve the National Kidney Foundation targets for these parameters. Studies are ongoing to determine the impact of the changes in these biochemical parameters on endpoints such as fractures, cardiovascular disease and mortality. Cinacalcet also shows efficacy in mild primary hyperparathyroidism, in which the level of expression of the receptor and the responsiveness of the pathological parathyroid gland(s) to Ca2+o are diminished, but has not yet received approval for this indication.
Calcium receptor antagonists, so-called calcilytics,(55) have been developed and their clinical utility is being explored, although they have not yet been approved for the treatment of any disorders involving the CaSR. In the presence of a calcilytic, a higher than usual calcium concentration is needed to suppress PTH levels to a given extent. As a result, the calcium receptor reads normocalcemia as hypocalcemia and secretes a pulse of PTH.(54),(55) If this pulse were of sufficient magnitude, it might enable the use of an orally administered calcilytic, instead of injected PTH, as an anabolic treatment for osteoporosis. Patients with activating mutations of the CaSR or activating anti-CaSR antibodies also represent potential therapeutic targets for the use of calcilytics, in that the sensitivity of the mutant or antibody-bound CaSR to Ca2+o could potentially be "reset" toward normal.(37)The Future of CaSR Research
There has been a great deal of progress in our understanding of the basic biology and molecular biology of the CaSR, as well as the molecular pathophysiology of the receptor in acquired and inherited disorders of parathyroid dysfunction. Areas in which we might anticipate further advances over the next decade are:
- greater knowledge of the receptor's roles in other tissues, particularly kidney, bone and intestine;
- further identification and characterization of inherited and acquired disorders of Ca2o-sensing, particularly outside of the parathyroid;
- wider application of CaSR-based therapeutics to parathyroid and non-parathyroid disorders.
Summary
Extracellular calcium (Ca2+o) is an important divalent mineral ion critical for numerous physiological processes. Critical insight into the regulation of Ca2+o was made about 15 years ago when the calcium-sensing receptor (CaSR), belonging to family C II of the super family of 7-transmembrane G protein-coupled receptors (GPCRs), was identified utilizing Ca2+o expression cloning technique. These efforts documented CaSR to be the sensor of Ca2+o in the parathyroid cell and, subsequently, other homeostatic tissues involved in Ca2+o homeostasis.
Since this time, there has been an explosion of information on the structure, ligands and binding properties of the CaSR, with a growing appreciation of aberrations of these properties that cause clinical disease. A newer field of in vivo CaSR biology has evolved with the development of mouse models using powerful tools of mouse genetics and molecular biology to determine whole animal and organ specific roles of the CaSR. Finally, the identification of binding sites, CaSR-specific antibodies and specific agonists and antagonists of the receptor have provided a foundation for the development of new chemical entities to treat diseases resulting from abnormalities of Ca2+o sensing by the CaSR.