Course Authors
Gregg L. Semenza, M.D., Ph.D.
Release Date: 09/15/2007
Upon completion of this Cyberounds®, you should be able to:
Discuss the molecular mechanisms by which changes in cellular PO2 are sensed and transduced to changes in gene expression through the activity of HIF-1
Discuss the role of HIF-1 and its downstream gene products in mediating adaptive responses to tissue ischemia and relevant translational research
Discuss the role of HIF-1 in the pathogenesis of cancer and relevant translational research.
Dr. Semenza will discuss the unapproved use of adenoviral HIF-1α/VP-16 gene therapy for critical limb ischemia.
Oxygen is essential for human survival. Complex circulatory and respiratory systems have evolved to continuously deliver adequate O2 to every cell in the body. The development of these systems and their proper utilization are dependent upon the activity of hypoxia-inducible factor 1 (HIF-1), a master regulator protein that controls the transcription of hundreds of genes according to changes in cellular PO2. Alterations in oxygen homeostasis play key roles in cancer and cardiovascular disease, leading to great interest in HIF-1, its downstream target genes and their protein products as novel therapeutic targets.
How Cells Sense and Respond to Changes in PO2
Every nucleated cell in the body is capable of sensing a reduction in oxygen concentration and responding by activation of HIF-1, which is composed of two subunits, HIF-1α and HIF-1β.(1) Under normoxic conditions, HIF-1α is degraded by the 26S proteasome, which is a multiprotein complex within the cell that serves to degrade regulatory proteins such as HIF-1α. By devoting considerable energy to the continuous synthesis and degradation of HIF-1α, the cell is poised to respond rapidly to any decrease in oxygenation. Under hypoxic conditions, the degradation of HIF-1α is inhibited and the protein rapidly accumulates and dimerizes with HIF-1β, which is constitutively expressed, to form an active transcription factor that can bind to DNA and activate gene transcription (Figure 1).
HIF-1α is targeted for proteasomal degradation when it is has been modified by a ubiquitin protein ligase, which is another multiprotein complex that catalyzes the covalent addition of the small protein ubiquitin to HIF-1α. The recognition of HIF-1α as a substrate for ubiquitination is mediated by binding of the von Hippel-Lindau tumor suppressor protein (VHL) to HIF-1α.(2),(3) VHL, in turn, only binds to HIF-1α that has been subjected to another post-translational modification, which is the hydroxylation of proline residue 402 or 564. This reaction is catalyzed by the prolyl hydroxylase PHD2, which utilizes O2 as a substrate. PHD2 enzymatic activity declines under hypoxic conditions. Thus, the half-life of HIF-1α is regulated through O2-dependent hydroxylation.
HIF-1 activates the transcription of genes that control adaptive responses to hypoxia. Many of these responses serve either to increase the delivery of O2 to cells or, alternatively, to allow cells to survive O2 deprivation. Among the many genes regulated by HIF-1 are: EPO and ADM, which encode erythropoietin and adrenomedullin, proteins that promote cell survival; VEGF and SDF1, which encode vascular endothelial growth factor and stromal-derived factor 1, proteins that promote angiogenesis; and GLUT1 and LDHA, which encode glucose transporter 1 and lactate dehydrogenase A, proteins that promote metabolic adaptation by increased utilization of glycolysis as a means of generating ATP (Figure 1). The role of these responses in the context of cardiovascular disease and cancer are discussed below.

Figure 1. Changes in Cellular O2 Concentration Are Transduced to the Nucleus as Changes in the Activity of HIF-1.
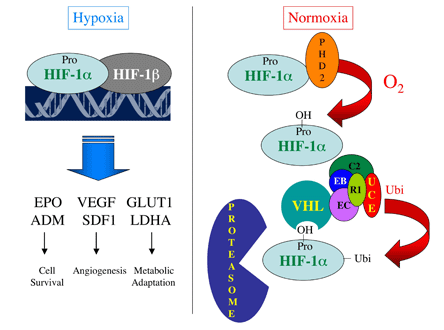
Right: Under normoxic conditions, HIF-1α is hydroxylated by PHD2, in a reaction that requires O2, and bound by VHL, which recruits a ubiquitin-protein ligase complex consisting of the proteins elongin C (EC), elongin B (EB), cullin 2 (C2), ring box protein 1 (R1) and a ubiquitin conjugating enzyme (UCE). Ubiquitinated HIF-1α is degraded by the proteasome.
Left: Under hypoxic conditions, PHD2 activity is inhibited, HIF-1α accumulates, dimerizes with HIF-1β, binds to DNA and activates the transcription of target genes whose protein products play critical roles in physiological responses to hypoxia.
HIF-1 Mediates Adaptive Vascular Responses to Ischemia
Three major processes by which blood vessels are formed and remodeled are referred to as vasculogenesis, angiogenesis and arteriogenesis.(4) Vasculogenesis denotes de novo blood vessel formation during embryogenesis, in which angiogenic progenitor cells migrate to sites of vascularization, differentiate into endothelial cells and coalesce to form the initial vascular plexus. The budding of new capillary branches from existing blood vessels is termed angiogenesis. Arteriogenesis refers to the remodeling of an existing artery to increase its luminal diameter in response to increased blood flow.
Stenosis of large conduit arteries in the heart or limbs from the presence of an atherosclerotic plaque results in decreased blood flow distal to the lesion. Impaired perfusion results in ischemia, a condition characterized by inadequate delivery of O2 and nutrients and inadequate removal of toxic metabolic wastes. In the femoral circulation, progressive stenosis leads to pain, loss of motor and sensory function, non-healing wounds and tissue necrosis that may eventually require limb amputation. New therapies are needed to promote limb salvage in patients with peripheral arterial disease. This is a particularly serious problem among diabetic individuals, who have a 20-fold increased risk of limb amputation compared to the general population.
Decreased perfusion downstream of an arterial stenosis results in hypoxia, an imbalance between O2 supply and demand, which is a physiological stimulus that induces cells to produce angiogenic growth factors/cytokines such as vascular endothelial growth factor (VEGF). These secreted proteins bind to cognate receptors (VEGFRs) on endothelial cells and activate signal transduction pathways that stimulate the cells to undergo sprouting angiogenesis. Thus, in order to satisfy this increased demand for O2, angiogenesis occurs to provide new capillary branches. VEGF is produced early in the angiogenic cascade and is responsible for initial activation of endothelial cells.(4),(5)
In addition to their ability to activate vascular endothelial cells within the ischemic tissue, certain angiogenic cytokines, such as VEGF, placental growth factor (PLGF) and stromal-derived growth factor 1 (SDF-1), stimulate the mobilization of a heterogeneous population of angiogenic cells from the bone marrow and other tissues (Figure 1), and their recruitment to sites of angiogenesis and arteriogenesis.(6),(7),(8) Although this process of angiogenic cell recruitment in response to ischemia is sometimes referred to as vasculogenesis, it appears to be quite different from the process that occurs during the formation of the initial embryonic vascular plexus. Among the important distinctions are the following:
- formation of the initial vascular plexus in the embryo is not driven by oxygen gradients;
- many of the cells recruited to sites of postnatal angiogenesis or arteriogenesis are inflammatory cells that are not directly incorporated into the new capillaries or remodeling arteries, respectively.
The identification of angiogenic growth factors led to clinical trials in which patients with myocardial or limb ischemia were administered the recombinant protein, e.g., VEGF, or a plasmid or viral vector encoding the protein. None of these trials demonstrated efficacy of the administered growth factor as compared to placebo.(9) Studies in animal models suggested that the combined effect of multiple angiogenic growth factors was critical to achieve a physiological vascular response such as placental PLGF and VEGF;(10) angiopoietin-1 (ANGPT1) and VEGF;(11) or platelet-derived growth factor (PDGF) BB and fibroblast growth factor 2.(12) Remarkably, HIF-1 functions as a master regulator by activating the transcription of genes encoding multiple angiogenic growth factors including VEGF, PLGF, PDGF-B, SDF1, ANGPT1 and ANGPT2. In animal models, injection of AdCA5, a replication-defective adenovirus that encodes an engineered form of HIF-1α that is resistant to degradation under normoxic conditions, has been shown to stimulate both angiogenesis and arteriogenesis.(13),(14)
HIF-1α Gene Therapy for Limb Ischemia: Preclinical/Phase I Studies
In a rabbit model of limb ischemia, the left femoral artery was occluded by the intravascular placement of titanium coils, resulting in a profound decrease in blood flow and blood pressure in the occluded limb. At the time of vessel occlusion, the rabbits were injected with adenovirus AdCA5, which encodes the constitutively active form of HIF-1α, or AdLacZ, which encodes β-galactosidase. Two weeks later, the AdCA5-treated animals showed a significantly increased recovery of perfusion, as demonstrated by arteriography and calf blood pressure measurements, and significantly increased arterial luminal diameter as demonstrated by immunohistochemistry.(14) Improved recovery of blood flow has also been demonstrated after injection of plasmid DNA encoding a chimeric HIF-1α/VP-16 protein into rabbits after surgical excision of the femoral artery(15) and a Phase I clinical trial was recently reported in which patients with critical limb ischemia were treated by intramuscular injection of a replication-defective recombinant adenovirus encoding the chimeric HIF-1α/VP-16 protein.(16)
Figure 2. Arteriography in Rabbits Subjected to Unilateral Femoral Artery Occlusion and AdCA5 Gene Therapy.
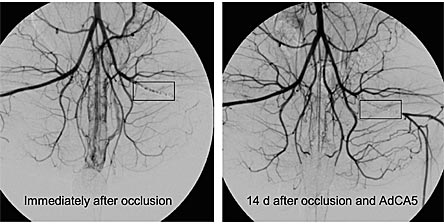
Left panel, the black rectangle indicates the location of the femoral artery occlusion due to intravascular placement of titanium coils. Note the absence of blood flow at the site of the coils and distal to them. Right panel, 14 days later, blood flow is still absent at the site of the coils, but distal to them blood flow has been reconstituted. The recovery of blood flow 14 days after occlusion results from the remodeling of pre-existing collateral vessels to increase their luminal diameter. Blood flow is proportional to (radius)3 so small increases in luminal diameter have major effects on blood flow. Adapted from (14)
Role of HIF-1 in Human Coronary Artery Collateralization
Angiographic studies of patients with ischemic heart disease secondary to atherosclerotic stenosis of a main coronary artery have revealed that, in approximately two thirds of patients, arteriogenesis has occurred to allow blood flow through a collateral vessel to perfuse myocardium downstream of the lesion. It is not known what genetic or environmental factors contribute to the absence of collateralization in one third of patients with significant coronary artery stenosis but these patients are more likely to die should they suffer a myocardial infarction. Analysis of a cytosine-to-thymine single nucleotide polymorphism in the HIF1A gene, which alters the sequence of HIF-1α protein from proline to serine at codon 582, revealed that the frequency of the rare T allele was increased 5-fold in patients without coronary collaterals (18.8%) as compared to patients with coronary collaterals (3.7%); in multivariate analyses, the presence of the CT or TT genotype was a significant negative predictor of collateral formation.(17)
These results suggest that genetic variation at the HIF1A locus may influence whether myocardial ischemia induces an adaptive vascular response (arteriogenesis) in patients with coronary artery disease. It remains to be determined whether patients carrying the rare allele have decreased HIF-1 activity and, if so, whether they might be particularly responsive to HIF-1α gene therapy as a means of stimulating coronary collateralization.
Erythropoietin Promotes Cell Survival
In patients with ischemic cardiovascular disease, an atherosclerotic plaque within a main coronary artery may rupture, resulting in sudden and complete obstruction to blood flow. Heart tissue distal to the occlusion will undergo infarction -- apoptotic and necrotic cell death -- from O2 deprivation. Infarct size is determined by many factors including the location and duration of the occlusion, the presence or absence of collateral vessels and many other factors.
Exposure of the heart to short episodes of ischemia protects against tissue injury following prolonged ischemia, a phenomenon known as preconditioning. Subjecting one organ to ischemia protects other organs in the same experimental animal, indicating the presence of circulating factors that protect against ischemic tissue injury. Protection of the heart against ischemia-reperfusion injury can also be induced by exposing mice to ambient hypoxia, which induces the HIF-1-dependent synthesis and secretion of EPO by the kidney.(18) Treatment of isolated hearts with recombinant human EPO (rhEPO) immediately prior to ischemia is sufficient to induce protection, demonstrating a direct effect of EPO on the heart.(19) rhEPO also protects against myocardial infarction when administered immediately following in situ coronary artery ligation.(20),(21),(22) rhEPO has also been shown to improve post-resuscitation myocardial dysfunction and survival in a rat model of cardiac arrest.(23)
EPO was originally identified as the hormone that controls red blood cell production by preventing the death of bone marrow erythroid progenitors and rhEPO has been used clinically in the treatment of anemia associated with chronic renal failure. However, recent studies in animal models such as those described above have demonstrated that pharmacologic administration of rhEPO results in improved recovery of many different tissues from many different types of tissue injury.(24)
A clinical trial of single-bolus administration of long-acting EPO to patients with acute myocardial infarction was recently reported.(25) A protective effect of EPO has also been reported in animal models of cerebral ischemia(26) and in a small double-blind study of stroke patients.(27) The slow pace of clinical trials of rhEPO administration to patients with acute myocardial infarction or stroke is puzzling given the dramatic results that have been obtained in animal models.
HIF-1 and Cancer Pathogenesis
In cancers, the O2 concentration is significantly reduced compared to surrounding normal tissue.(29) As a result, HIF-1 activity is induced both within hypoxic cancer cells as well as within hypoxic stromal cells. In addition to the physiological induction of HIF-1 activity in response to hypoxia, mutational mechanisms have been identified in cancer cells that lead to the loss of O2-dependent degradation of HIF-1α. The most dramatic of these is the effect of von Hippel-Lindau tumor suppressor protein (VHL) loss-of-function in clear cell renal carcinoma. As described above, VHL recognizes HIF-1α that has been hydroxylated on proline residue 403 and/or 564. The binding of VHL leads to the ubiquitination and proteasomal degradation of HIF-1α in well oxygenated cells (Figure 1). Loss of VHL leads to constitutively high levels of HIF-1α (Figure 3) and dysregulated expression of HIF-1 target genes, including those encoding the angiogenic cytokines SDF-1 and VEGF, resulting in tumors that are highly vascularized.(30),(31)
Immunohistochemical analysis of biopsy sections (Figure 3) has established significant associations between HIF-1α overexpression and patient mortality in a wide variety of human cancers.(1) The molecular pathobiology underlying these clinical associations has been investigated in cell culture and tumor xenograft models and has revealed roles for HIF-1 in virtually every critical aspect of cancer biology, as detailed below.
Figure 3. Immunohistochemical Detection of HIF-1α Protein within Cancer Cell Nuclei in a Biopsy Section Taken from a Renal Cell Carcinoma of the Clear Cell Type.
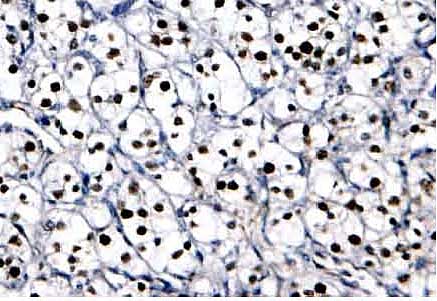
In addition to its characteristic histological appearance, the signature lesion in this cancer is VHL loss of function which results in high levels of HIF-1α, as evidenced by the brown staining of every tumor cell nucleus. Adapted from (29)
Immortalization
Immortalization of human cancer cells involves transcriptional activation of the TERT gene encoding telomerase. HIF-1 has been shown to activate TERT transcription by binding to a site in the promoter of the gene.(31),(32)
Maintenance of Stem Cells
The role of stem cells in cancer pathogenesis has received renewed attention recently.(33) Hypoxia and HIF-1 regulate several proteins that play important roles in stem cell biology, including BCRP, Notch and Oct4.(34)
Genetic Instability
Cancer cells have impaired capability to repair damaged DNA.(35) The DNA mismatch repair system safeguards genomic integrity by correcting DNA replication errors and preventing recombination between non-homologous DNA. The mismatch repair protein MutSα is a heterodimer composed of the products of the MSH2 and MSH6 genes, which are transcriptionally activated by a complex consisting of the transcription factors SP1, C-MYC and P53 under non-hypoxic (low HIF-1α) conditions.(36) Under hypoxic conditions, HIF-1α is induced and displaces C-MYC and P53 from interaction with SP1 and thereby blocks transcriptional activation of the MSH2 and MSH6 genes. This is a unique function of HIF-1α as it does not require dimerization with HIF-1β or direct binding to DNA.
Angiogenesis
As described above, HIF-1 regulates the expression of genes encoding multiple angiogenic growth factors including ANGPT1, ANGPT2, PLGF, PDGF-B, SDF-1 and VEGF. Genetic manipulation of human colon cancer cells to increase HIF-1α levels results in increased VEGF production and increased tumor xenograft growth and angiogenesis.(37) Conversely, genetic manipulation of human gastric cancer cells to decrease HIF-1 activity results in decreased VEGF production, decreased tumor xenograft growth and decreased tumor vessel area that is associated with defective vessel maturation.(38) Tumor vascularization is also impaired in mice lacking HIF-1α in endothelial cells.(39)
Glucose and Energy Metabolism
Carcinomas have increased rates of glycolytic metabolism even under non-hypoxic conditions, which is referred to as the Warburg phenomenon.(40) HIF-1 activates the transcription of the genes that encode glucose transporter 1 and glucose transporter 3, as well as the glycolytic enzymes hexokinase 1, hexokinase 2, phosphofructokinase L, aldolase A, aldolase C, glyceraldehydephosphate dehydrogenase, phosphoglycerate kinase 1, phosphoglyceromutase, enolase 1, pyruvate kinase M and lactate dehydrogenase A (LDHA).(41) In human breast cancer cell lines, there is a strong correlation between HIF-1α levels and glucose consumption, suggesting that dysregulated HIF-1α expression contributes to the Warburg phenomenon.(42) In addition, HIF-1 activates the transcription of the PDK1 gene encoding pyruvate dehydrogenase (PDH) kinase 1, which phosphorylates and inactivates PDH, the enzyme that converts pyruvate to acetyl coenzyme A for entry into the mitochondrial tricarboxylic acid cycle. Together with LDHA, PDK1 shunts pyruvate away from the mitochondria through its conversion to lactate. In VHL-deficient renal carcinoma cells, the biogenesis of mitochondria is actively inhibited by HIF-1, thus further tipping the balance in favor of glycolytic rather than oxidative metabolism.(43)
Autocrine Growth Factor Signaling
Tumor cells survive and proliferate in an uncontrolled manner by establishing autocrine signaling pathways in which they express both the growth factor and its cognate receptor. Colon cancer cells express insulin-like growth factor 2 (IGF-2) and its receptor, IGF-1R. Stimulation of IGF-1R tyrosine kinase activity induces HIF-1α expression in colon cancer cells through activation of the phosphatidylinositol 3-kinase (PI3K) and MAP kinase pathways.(44) In addition, IGF2 is a HIF-1-regulated gene.(45) Thus, increased HIF-1 activity will lead to increased IGF-2 production, which will lead to increased IGF-1R tyrosine kinase activity and activation of the PI3K and MAP kinase pathways that control cell survival and proliferation. A similar autocrine loop exists in renal cancer cells, which express both the epidermal growth factor receptor and its ligand transforming growth factor-α, which is encoded by a HIF-1-regulated gene.(46),(47)
Invasion and Metastasis
Invasion through the basement membrane that delimits the normal epithelium is the defining property of the cancer cell and is the property that is most responsible for patient mortality. Invasion requires transformation from an epithelial cell phenotype, which is characterized by a rigid cytoskeleton and strong cell-cell interactions, to a mesenchymal phenotype, which is characterized by a fluid cytoskeleton and reduced cell-cell interactions (Figure 4). HIF-1 regulates the expression of genes encoding vimentin and several keratins that are upregulated in cancer cells.(47) The loss of E-cadherin expression is associated with the epithelial-to-mesenchymal transformation in most solid tumors and in VHL-deficient renal cell carcinoma HIF-1 mediates repression of E-cadherin gene expression.(48),(49) Invasion also requires digestion of the extracellular matrix through the activity of matrix metalloproteinases (MMPs), cathepsins and the urokinase plasminogen activator-UPA receptor (UPAR) system. HIF-1 regulates the expression of genes encoding cathepsin D, membrane type-1 matrix metalloproteinase (MMP), MMP-2, urokinase plasminogen activator receptor (uPAR) and the proprotein convertase furin.(47)
Several factors have been shown to stimulate the motility of cancer cells, thus promoting invasion and metastasis, most notably C-MET, which is regulated by HIF-1.(50) Expression of the chemokine receptor CXCR4 has been implicated in the homing of metastatic breast cancer to lung, which expresses high levels of the ligand stromal-derived factor-1.(51) CXCR4 is over-expressed in VHL-deficient renal carcinoma cells as a result of dysregulated HIF-1 activity.(52) In breast cancer, HIF-1 promotes both osteolytic bone metastases(53) and pulmonary metastases.(53)
Figure 4. Involvement of HIF-1 in Tumor Cell Invasion.
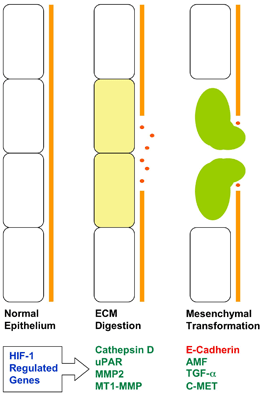
Gene transcription that is activated (green) or repressed (red) by HIF-1 is shown.
Resistance to Therapy
In an analysis of oropharyngeal cancer biopsies, patients whose tumors had the highest levels of HIF-1α expression (>10% of cells in the biopsy) had a 3-fold increased risk of failure to achieve a complete remission after radiation therapy.(55) HIF-1-deficient tumor xenografts manifest increased susceptibility to radiation as well as to carboplatin and etoposide, which are chemotherapeutic agents that, like radiation, induce double-stranded DNA breaks.(56),(57)
Radiation of tumor xenografts induces HIF-1 activity in cancer cells leading to the secretion of VEGF and fibroblast growth factor 2, which serve as survival factors for tumor blood vessels,(59) thus providing a mechanism by which HIF-1 may promote radiation resistance. This protective effect of HIF-1 is overcome when radiation is combined with the anti-angiogenic agent canstatin.(59) Chemotherapy resistance can be induced by upregulation of genes encoding multidrug transporters. HIF-1 activates transcription of the ABCB1 and ABCG2 genes, which encode the MDR1 and BCRP multidrug transporters, respectively.(60),(61)
Conclusion
HIF-1 is required for normal development and physiological responses to hypoxia. In ischemic cardiovascular disease, gene therapy strategies designed to increase HIF-1 activity may be useful in promoting vascularization. In cancer, small molecule inhibitors of HIF-1 activity may be useful in combination with other chemotherapeutic agents. Clinical trials are currently underway to test each of these hypotheses.