Course Authors
Matthew H. Porteus, M.D., Ph.D.
Release Date: 09/10/2006
Upon completion of this Cyberounds®, you should be able to:
Describe the different types of viral vectors that are being used in gene therapy
Summarize the successes and setbacks in the recent clinical trials for gene therapy
Describe the future directions of gene therapy research to improve the safety of gene therapy.
One of the conundrum's of modern medicine is our increasing understanding of how many diseases are caused by either acquired or inherited mutations (genetic diseases) and our increasingly obvious inability to treat those diseases at that level. Our treatments are usually directed at the consequence of mutations rather than directly at the genetic level.
The monogenic diseases, of which there are thousands, are caused by an inherited mutation in a single gene. Some examples of monogenic diseases are sickle cell disease (caused by mutations in both copies of the β-globin gene), cystic fibrosis (caused by mutations in both copies of the CFTR gene) and Huntington's diseases (caused by a specific type of a mutation in one copy of the Htg gene). Monogenic diseases permeate all medical specialties. Cancer is a second type of genetic disease. In cancer, certain cells have acquired mutations that transform the cell from a normal cell into a cancer cell.
In the early 1970s scientists discovered how to cut and paste different pieces of DNA together, thereby creating recombinant DNA technology. Using recombinant DNA technology, scientists could modify genomes in model systems and thereby create cells and organisms with new phenotypes. Sometimes these new phenotypes were designed to mimic human diseases and thus create models for human disease. Other times these experiments showed that recombinant DNA could be used to "fix" a phenotype that resembled a human disease. From these types of experiments, the field of gene therapy was born: the manipulation of the nucleic acid content of a cell, usually the genome directly, for therapeutic benefit.
Gene therapy is being developed for a number of purposes, including the possibility of treating monogenic diseases and cancer. While the focus here is on gene therapy for monogenic diseases, in the next section I will briefly discuss the use of gene therapy for cancer.
Gene Therapy for Cancer
There are a number of different approaches to using gene therapy to treat cancer. I will briefly discuss three:
- the use of oncolytic viruses;
- using gene therapy to modify the cancer cell;
- using gene therapy to modify the host immune response.
One of the characteristics of epithelial carcinomas is that they have acquired mutations in tumor suppressor genes. Fifty percent of carcinomas, for example, have mutations in p53. Researchers have developed viruses, such as a modified adenovirus, that require the absence of p53 in order to replicate. Thus, infecting normal cells with these "oncolytic" viruses will have no effect because the virus will be unable to replicate.(2) But if the virus infects a cell that is p53 deficient, such as a tumor cell, it will replicate, lyse the cell and be released to infect other cells. In this way, the virus will preferentially kill the tumor cells while preserving normal cells.
The second way gene therapy is being studied to treat cancer is to use viral vectors to deliver new genes to the cancer cell so that it adopts a new phenotype. This new phenotype would serve to activate the host immune system against both the infected and non-infected tumor cells. For example, in melanoma, researchers have infected melanoma cells with a recombinant retrovirus that expresses the GM-CSF, a cytokine. These GM-CSF-expressing melanoma cells serve to activate the host immune response against the melanoma cells in general such that the immune system destroys both the GM-CSF-expressing melanoma cells and the non-GM-CSF-expressing melanoma cells. In xenograft mouse models, this strategy was successful in eliminating tumors. In human trials, this strategy did result in an inflammatory infiltrate around the melanoma but did not produce any clinical benefit.(3)
The third gene therapy approach has been to activate the host immune system against the tumor either by using tumor vaccines or by modifying host T-cell receptors so that they recognize tumor antigens.(4)
General Strategies for Gene Therapy of Monogenic Diseases
Monogenic diseases can be thought of as a car with broken headlights. In a recessive monogenic disease, both headlights are broken. In a dominant monogenic disease, only one headlight is broken but that is sufficient to cause disease. It can cause disease because the driving conditions are such that a single functional headlight is not adequate (haplo-insufficiency) or it could be actively deleterious, as in Huntington's disease (such as by pointing directly in the driver's face, thereby blinding the driver).
There are two general strategies for gene therapy for monogenic diseases: gene addition and gene correction (Figure 1).

Figure 1. Comparison of Gene Addition vs. Gene Correction.
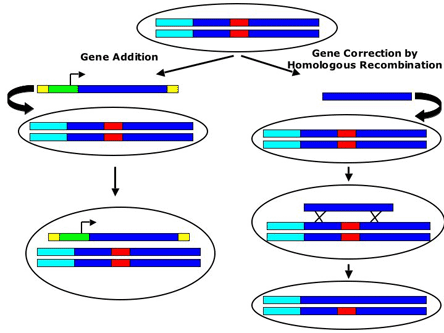
This figure schematizes the difference between gene addition and gene correction. The dark blue bars represent the chromosomal gene of interest. The red box represents the disease causing mutation for an autosomal recessive disease. The light blue box represents the endogenous regulatory region (promoter and enhancers) for the gene. In gene addition (on the left), an additional copy of the gene under the control of a heterologous promoter (green box) is introduced into the cell through virus (the yellow boxes represent the ITR's that cap the end of the virus). This transgene is then maintained as a third copy of the gene in the cell (the other two copies persist but remain mutated). In gene correction (on the right), a fragment of the mutated gene that does not contain the mutation is introduced into the cell. This fragment then replaces the corresponding fragment in the endogenous gene by homologous recombination (represented by the two "X's"). The fragment is eventually lost by either degradation or dilution from cell division and in the end the cell is left with the two endogenous copies of the gene but one has been corrected and no longer contains the mutation (no red box).
In gene addition, an extra gene is added into the genome. This extra wild-type gene is then expressed, hopefully providing enough of the wild-type protein product to ameliorate the disease phenotype. Usually the gene is introduced into the genome using viral vectors and integrates into the genome in an uncontrolled fashion. Gene addition is like fixing the car with two broken headlights by attaching a third headlight to the car. In gene addition we do not control where the headlight is attached; sometimes the headlight is non-functional, sometimes the headlight is functional and occasionally the headlight is deleterious. Gene addition, adding a third headlight, will not work for genetically dominant diseases, such as Huntington's disease, where one broken headlight is actively causing harm. Some people use the term "gene replacement" to describe gene addition but this term is misleading as it suggests that the new gene is replacing the mutated gene. Gene addition has been the focus of a majority of gene therapy research. In the next sections I will discuss the different viral vectors that are used in gene addition type gene therapy and the results of recent clinical trials for monogenic diseases using those viral vectors.
An alternative approach is to use gene correction to figure out a way to fix one or both of the broken headlights. Conceptually, fixing the broken headlights seems like the best way of doing gene therapy. One approach to fixing the headlight is to use homologous recombination to directly correct the disease-causing mutation. The use of homologous recombination for gene therapy will be discussed in the section on "Future Directions."
Viral Vectors for Gene Therapy
In gene addition gene therapy, the gene of interest ("transgene") needs to be introduced into a cell. For monogenic diseases the transgene needs to be stably expressed. Stable expression requires two preconditions: that the vector remains in the cell and the promoter driving transgene expression remains active.
In non-dividing cells, the vector can remain in the cell either episomally or integrated into the genome. In dividing cells, the vector is stable in the cell either if it integrates into the genome, and thus is replicated along with the host genome during the cell cycle, or if it contains an element that allows it to be replicated episomally (as can occur with vectors based on the Epstein-Barr virus backbone).
One of the critical aspects of all gene therapy is to be able to deliver the transgene into the cell of interest. In cell lines that grow in culture, physical transfection techniques using cationic lipids or electroporation are efficient ways of delivering genes into cells. But most primary cells are relatively resistant to these physical methods of transfection. An alternative approach is to use recombinant viruses to deliver genes into cells. Viruses have evolved to be able to enter cells efficiently and deliver genes into the cell (infection). By removing the viral genes and replacing them with transgenes, scientists have created recombinant viruses that have co-opted the ability of viruses to infect cells and use them as efficient ways to deliver genes into cells (gene transfer). Figure 2 shows how a natural virus is transformed into a recombinant virus that then can be used as a vector to deliver genes into cells.
Figure 2. Creating Recombinant Viral Vectors for Gene Therapy.
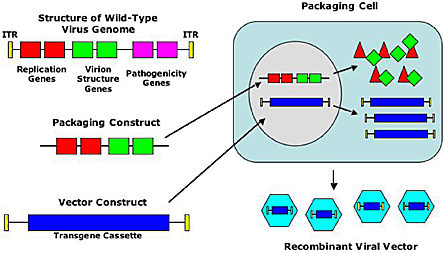
Recombinant viral vectors are made by deleting the natural genes in the virus (labeled in red for replication, green for virion structure and purple in for pathogenicity) and replaced with a "transgene cassette" (in blue). The transgene cassette contains the various DNA elements that the investigator chooses. Sometimes the cassette contains a promoter but in retroviral vectors the transgene promoter can be derived from the ITR ("inverted terminal repeats"). In addition to acting as a promoter, the ITR's also serve as replication origins in the production of the virus and packaging signals to incorporate the viral DNA into the viral capsid. The vector construct is converted to recombinant viral vector by co-transfecting a packaging cell line (often HEK-293 cells) with the "vector construct" and the "packaging construct." The packaging construct provides the gene products in trans necessary to package the vector construct into a viral particle creating the "recombinant viral vector." The recombinant viral vector can then be purified and used to infect target cells of interest to deliver the transgene cassette to that cell.
There are a number of different viruses that have been modified for use as gene therapy vectors.(1) I will briefly discuss four of these: adenovirus, adeno-associated virus, retrovirus and lentivirus.
Adenovirus
Adenoviruses are large (35kB), double-stranded DNA viruses that are a major cause of upper respiratory tract infections in humans but can cause infections of multiple other tissues. They do not contain elements that promote integration of the vector genome into the cell and most stay episomal. Recombinant adenoviral vectors have been deleted for the elements that allow them to replicate. In dividing cells, therefore, the adenoviral genome will be lost. Thus, adenoviral vectors are good vectors for non-dividing cells such as quiescent hepatocytes. The major drawback of adenoviral vectors is that most humans have been infected with many different adenoviruses and have developed immunity to them. Thus, the administration of recombinant adenoviral vectors that have been deleted of many of the natural viral gene products can still provoke an inflammatory response. Adenoviral vectors have been used in a number of cancer gene therapy trials and in the gene therapy trial for ornithine transcarbamylase deficiency.
Adeno-associated Virus (AAV)
Adeno-associated virus is a single-stranded DNA virus from the Parvovirus family. It is unable to replicate without the help of co-viral infection (either by adenovirus or herpes simplex virus). After infection, recombinant AAV (rAAV) viral vectors can either be maintained episomally, integrate randomly in the genome or integrate into the genome via homologous recombination. While other Parvovirus family members, such as Parvovirus B19, can cause disease in humans, adeno-associated virus is believed to be non-pathogenic in humans. There are a number of serotypes of AAV that differ in their cell surface receptor and ability to infect different cell types.
A disadvantage of rAAV vectors is that they can only package 4.7 kb of DNA. This amount of DNA limits the complexity of regulatory elements or size of the transgene that can be carried by rAAV. Like adenovirus, humans often have been exposed to different AAV infections and have pre-existing humoral immunity to certain serotypes. Thus, different serotypes have the advantage of not only being able to infect different cell types but also may be less prone to rapid clearance by the human immune system. Recombinant AAV viruses have been used in clinical gene therapy trials for the treatment of hemophilia B (Factor IX deficiency).
Retrovirus
Retroviruses are single-stranded RNA viruses that can infect a wide variety of cell types. After infection of the cell, retroviruses use reverse transcriptase to convert the single-stranded RNA genome into a double-stranded DNA genome and then use a virally encoded integrase to integrate the double-stranded DNA genome into the genomic DNA of the host cell. Retroviruses are "integrating" viruses and do not stay episomal. Once integrated into the host genome, the retroviral DNA replicate along with the host genome. Retroviral vectors used in gene therapy are replication incompetent so they cannot create more viral particles. Initially retroviruses were thought to integrate randomly into the genome but recent studies have shown that retroviruses preferentially integrate into the 10 kB upstream regulatory regions of genes. A disadvantage of conventional retroviruses is that they do not efficiently infect non-dividing cells because they do not cross the nuclear membrane. Thus, retroviruses can be best used to infect dividing cells. Retroviruses have been employed in clinical gene therapy trials for severe combined immunodeficiency and chronic granulomatous disease.
Lentivirus
Lentiviruses are a sub-family of retroviruses that are single-stranded RNA viruses that infect a wide variety of cell types including non-dividing cells. Recombinant lentiviral vectors have been derived from the HIV virus. In recombinant lentiviral vectors, all of the HIV genes have been replaced with DNA of the investigator's choice, thus making the resulting virus replication incompetent and non-pathogenic. Lentiviral vectors do not integrate randomly into the host genome. Instead, they integrate preferentially into the coding regions of actively expressed genes. Lentiviral vectors are undergoing extensive pre-clinical testing for clinical gene therapy trials but have not yet been used in a human trial.
Promoters to Drive Gene Expression
One of the important issues in gene therapy is how to express the introduced wild-type gene. There are several different options to express the introduced gene. The first is to use viral elements to drive expression of the transgene. For example, in retroviral vectors, the long-terminal repeat (LTR) at the ends of the viral genome not only serve as packaging signals but can also function as promoters/enhancers. Thus, many investigators have used these natural viral elements to drive expression of the transgene. The problem with this approach is that the level of expression is controlled by a viral element and that level may or may not be appropriate for the clinical disease. A further problem is that just as viruses have evolved to infect and replicate in a wide variety of cells, cells have evolved a number of ways to suppress viral infection and replication. In addition to the fascinating Apobec family of proteins which seem to be an intra-cellular method of immunity against viral infection,(5) cells have also developed ways to shut down the expression of viral promoters, including viral LTRs, and thereby shut down the expression of viral genes. Thus, using viral elements to drive expression of the transgene can be problematic.
A second approach is to use promoters/enhancers derived from mammalian genes that are ubiquitously expressed, such as the elongation-factor 1 alpha and ubiquitin promoter. While these promoters have a decreased tendency to be "silenced," they also suffer from the problem that they may not express the transgene at the precise level necessary.
A third approach is to use tissue specific promoters/enhancers. These elements have the advantage of giving expression in specific cell types and can be modified to give different levels of expression. The disadvantage of such promoters is that they are usually more complex and require more study to determine precisely which elements are needed to give the appropriate levels of expression.
The problem of position effect variegation (PVE) affects all gene therapy approaches that require the integration of the transgene. In PVE, the expression level of the transgene is modified by the local milieu in which it integrates. For example, in some case the transgene may integrate near a strong enhancer and be expressed at higher than expected levels. More problematically, in other cases the transgene may integrate in a region of the genome that is prone to undergo chromatin modifications that silence gene expression, such as heterochromatin. In this case, the transgene would be expressed at much lower levels than expected. One potential solution to the problem of position effect variegation is to incorporate insulator elements into the gene delivery vector. I will discuss insulators in further detail below.
One of the advantages of using gene correction as gene therapy is that one does not have to worry about promoter or expression issues. If one can correct the endogenous gene in the correct cell type, then the endogenous promoter/enhancer elements will generate sustained and appropriate levels of expression.
Gene Therapy Clinical Trials
In the next section, I will discuss the clinical gene therapy trials for five different monogenic diseases: ornithine transcarbamylase deficiency (OTC deficiency), hemophilia B (Factor IX deficiency), X-linked severe combined immunodeficiency (SCID-Xl), severe combined immunodeficiency from adenosine deaminase deficiency (ADA-SCID) and chronic granulomatous disease (CGD). Each of these trials highlights certain successes and setbacks in the continued development of gene therapy.
Onithine Transcarbamylase (OTC) Deficiency
OTC deficiency is an autosomal recessive monogenic disease caused by a deficiency in the OTC enzyme. There are variable forms of OTC deficiency including a severe form that results in infant death and can only be cured by orthotopic liver transplantation and milder forms that can be controlled by diet and dietary supplements. The curative nature of liver transplantation suggested that gene transfer of the OTC gene to hepatocytes might be able to cure the disease.(6),(7) In the ethical discussion preceding this trial, it was decided that infants with the severe form of the disease would not be eligible because there was no alternative therapy for these infants and the parents would have a degree of desperation that would preclude them from giving true informed consent. Thus, only patients with mild forms of the disease were eligible.
The trial consisted of administering 2 x 109-6 x 1011 viral particles per kilogram of recombinant adenoviral vector expressing the OTC gene into the hepatic artery. The goal was to infect the maximal number of hepatocytes. Eighteen patients were treated with escalating doses of recombinant adenoviral vector. Several patients had a transient increase in their plasma OTC levels but no sustained response. Several patients had a transient elevation in their hepatic enzymes suggesting a transient hepatic inflammation but they recovered without further events. The trial ended, however, when a 19-year-old patient developed a severe inflammatory reaction after the infusion of the recombinant adenoviral vector, presumably from an immune system that was activated to respond to adenovirus from prior infections, that resulted in his death. The activation of a lethal inflammatory response from a primed immune system highlights a major problem with the in vivo administration of viral vectors. Moreover, since we currently have no way of predicting which patients are primed in this way, the in vivo administration of adenoviral vectors can now be seen as analogous to a high tech form of Russian roulette.
Hemophilia B (Factor IX Deficiency)
Hemophilia B is an X-linked recessive disorder caused by mutations in the Factor IX gene. Factor IX is an integral component of the clotting cascade and males with deficiency in factor IX are prone to recurrent and debilitating joint, muscle and soft-tissue bleeds. The treatment of hemophilia B is either prophylactic infusions of recombinant or virally inactivated factor IX and/or infusions of factor IX in response to active bleeds. The severity of hemophilia B can be greatly reduced by just a small amount of factor IX. For example, patients with 1-5% factor IX have much less severe disease than those who have no circulating factor IX and patients with 5% or greater factor IX levels can be symptom free. Thus, a small amount of circulating factor IX could have a profound clinical impact.
Two gene therapy trials for hemophilia B were initiated using rAAV to deliver the factor IX transgene. In the first trial, rAAV was injected into the muscle of eight patients.(8) These patients had no rise in their circulating factor IX levels but biopsy of the muscle injection sites did show gene transfer into the muscle cells and expression of the factor IX gene. In the second trial, 8 x 1010-2 x 1012 viral particles per kilogram of recombinant AAV vector were injected into the hepatic artery of seven adult male patients.(9) Two patients had factor IX transiently in their blood but none had sustained significant circulating factor IX levels. Subsequent analysis showed that transduced hepatocytes were eliminated by the immune system, probably because of prior immunity to the AAV serotype used. In addition, in this trial, semen was examined for evidence of transduction of germ line cells and was found in the seminal fluid. Careful separation of the components of the seminal fluid showed that the rAAV vector came from the fluid bathing the sperm and not from the sperm itself showing that transduction of germ line cells had not occurred. Nonetheless, it raised the concern that in vivo administration of viral vectors might have transduced germ line cells.
X-Linked Severe Combined Immunodeficiency (SCID-Xl)
SCID-Xl is an X-linked recessive disease caused by mutations in the interleukin-2 receptor common gamma chain (IL2RG) gene. Patients have no functional T or B-cells and without treatment patients will usually die in infancy from overwhelming viral infections. SCID-Xl can be cured by bone marrow transplantation. If the donor is an immunologically matched sibling without the disease, the success of bone marrow transplantation is 90%. If the patient does not have a sibling-matched donor, then haploidentical transplantation can be done using one of the parents as a donor. The success of haploidentical transplantation is about 60%.
In 2000 Fischer and his colleagues reported on a boy with SCID-Xl who spontaneously developed a functional immune system that protected him from viral infections.(10) They found that a single precursor cell had undergone a somatic reversion event (a cell that originally carried the inherited mutation acquired a somatic mutation that changed the mutation back to wild-type). This unique event highlighted that significant clinical benefit might be gained even if only a few precursor cells underwent gene transfer. This patient highlights a phenomenon called "selective advantage" in which cells that have a functional IL2RG gene will have a growth advantage over cells that do not. If cells having undergone gene transfer have a selective advantage, as they would for SCID-Xl, it dramatically increases the probability of success for the gene therapy protocol.
With the development of protocols to infect human CD34+ cells (the population that contains hematopoietic stem cells) using retroviral vectors, the Fischer group embarked on clinical gene therapy trial for SCID-Xl, the "French trial." They purified CD34+ cells from 10 patients and retrovirally infected the purified cells ex vivo with a vector that expressed the wild-type IL2RG gene from the retroviral LTR and infused the autologous cells back into the patients without giving prior myeloablative therapy.(11),(12) Of the 10 patients, nine developed a functional immune system. Six of the patients have remained alive and well without any serious adverse events.
Unfortunately, two to three years after their gene therapy procedure, three patients developed a T-cell leukemia. Two of those patients remain alive but one has passed away. Molecular analysis of all three leukemias showed that a process called "insertional oncogenesis" had occurred. Insertional oncogenesis occurs when a vector insertion activates an oncogene, thereby transforming the normal cell into a cancer cell. In this SCID-Xl trial, all three leukemias were caused by the insertional activation of the LMO-2 proto-oncogene by the retroviral LTR. This trial is currently halted as the investigators study ways to minimize the risk of insertional oncogenesis.
A second gene therapy trial, the "British trial," for SCID-Xl is currently open and run through the Great Ormond St. Hospital in London. Like the French trial, the British trial is using a retrovirus expressing the wild-type IL2RG gene from the viral LTR to infect purified patient CD34+ cells. They have published the reports on four patients.(13) All four patients have developed a functional immune system and there have been no serious adverse events. When this study was published, the patients had not reached the critical two- to three-year period after infusion when the patients in the French trial developed leukemia. Thus, the gene therapy community is awaiting the follow-up on these patients as they pass through this seemingly critical period. In the meantime, since the British have not had any serious adverse events, this trial is continuing to enroll patients.
Severe Combined Immunodeficiency from Adenosine Deaminase Deficiency (ADA-SCID)
ADA-SCID is an autosomal recessive disease caused by mutations in the adenosine deaminase (ADA) gene. Mutations in this gene disrupt the ability of cells to utilize the salvage pathway of nucleotide biosynthesis. Developing lymphocytes are particularly sensitive to perturbations in this pathway and undergo apoptosis. Consequently, patients lack functional T- and B-cells. Without treatment, patients with ADA-SCID die in infancy, usually from overwhelming viral infections. Patients with ADA-SCID can either be treated by matched sibling bone marrow transplantation or by regular injections of adenosine deaminase enzyme conjugated to polyethylene glycol (PEG-ADA). These injections are effective but very expensive. Patients with ADA-SCID have undergone several gene therapy trials in the United States, none of which resulted in clinical benefit to the patients. In 2002, Aiuti et al. (the "Italian trial") reported on their gene therapy trial for ADA-SCID.(14) In this trial, they infected purified CD34+ bone marrow cells with a retrovirus expressing the ADA gene from the viral LTR and then infused these infected cells into the patient. Prior to the infusion of the infected cells, however, they gave the patients a moderate but non-myeloablative dose of busulfan to facilitate engraftment of the transduced CD34+ cells. The two patients reported have developed functional immune systems without the report of any serious adverse events. This trial continues to enroll patients.
Chronic Granulomatous Disease (CGD)
CGD is caused by mutations in genes that are part of the complex that generates the respiratory burst in neutrophils which enables neutrophils to kill micro-organisms. There are both autosomal recessive and X-linked recessive forms of the disease. In both forms, patients have abundant neutrophils but these neutrophils are unable to effectively kill bacteria and fungi. These patients develop life-threatening bacterial and fungal infections and without appropriate treatment would die in early childhood. Like other monogenic hematopoietic diseases, CGD can be cured by bone marrow transplantation. If the patient does not have an immunologically matched sibling donor without CGD, then standard therapy consists of regular injections of interferon, prophylactic antibiotics such as bactrim, prophylactic antifungals such as itraconazole and aggressive management of acute infections.
In 2006, Ott et al. published the results of two patients with X-linked CGD who underwent gene therapy.(15) In this work, the investigators purified CD34+ bone marrow cells and infected them with a retrovirus that expressed the gp91phox gene from the retroviral LTR. They then infused these transduced cells into the patients after giving a moderate dose of busulfan (8 mg/kg) to enhance engraftment of the transduced cells (just as the Italians did in their ADA-SCID trial). Both patients developed a sufficient number of neutrophils with enough respiratory burst activity to eliminate chronic bacterial and fungal infections. Both patients also developed a clonal myeloproliferative condition by the insertional activation of the MDS-Evi1 gene. The Evi1 gene is a proto-oncogene and its activation is associated with the occurrence of myeloid leukemias.(16) Thus, just as in the French SCID-Xl trial, the German CGD trial was associated with the activation of a proto-oncogene by the nearby insertion of the retrovirus and the subsequent activation of the proto-oncogene by the retroviral LTR.
Since the publication of this article, the first patient has died. The cause of death has been reported to be from overwhelming sepsis from a ruptured intestinal abscess. The final report is pending but it seems likely that the patient did not die directly from an adverse event from the gene therapy protocol but may have died from lack of sustained efficacy of the protocol. The second patient remains alive, without evidence of transformation of his myeloproliferative state into a frank leukemia. This trial is currently on hold.
Future Directions in Gene Therapy
Depending on one's personality, one can interpret the clinical gene therapy trials as showing that the glass is half-empty or half-full. Clearly, there have been examples of lack of efficacy and unexpected serious adverse events (the glass is half-empty). But there are now an increasing number of patients (perhaps over 20 worldwide) who have had significant improvements in the quality of their lives after undergoing gene therapy (the six patients in the French trial, the patients in the on-going British SCID-Xl trial and the patients in the on-going Italian ADA-SCID trial) (the glass is half-full). Whether you see the glass as half-full or half-empty, it is clear that there is room for improvement. In the following, I will discuss some of the approaches that are being taken to improve gene therapy.
Avoiding the Immune Response
The OTC and hemophilia trials both showed that the immune response is a powerful limitation to in vivo gene therapy protocols. In the OTC trial, the overwhelming activation of the immune response resulted in the death of a patient and, in the hemophilia trial, the elimination of transduced cells by the immune response abrogated any potential clinical benefit but fortunately did not result in any serious adverse events. Designing ways to minimize the immune response is an important area of research and there are several strategies.
One solution is to avoid in vivo administration of viral vectors entirely and use ex vivo strategies only. In this way, the immune system is never exposed to the antigenic stimuli from the viral vector. Such ex vivo strategies depends on being able to remove a large number of cells from the patient, manipulate them outside the body, re-infuse them back into the body and then have them migrate back to where they belong. These are characteristics of hematopoietic stem cells but for many other tissue types and stem cell types this approach is currently not feasible. Thus, the study of how to identify, grow, and re-infuse tissue specific stem cells from non-hematopoietic sources is an important area of research.
A second solution is to transiently immunosuppress the patient during the administration of the viral vector. This will probably require both blocking pre-existing B-cell humoral immunity and suppressing the adaptive T-cell response until the viral antigens are no longer present. Currently, researchers are actively studying the immune response to gene therapy vectors and devising potential ways of suppressing that response. While long-term immunosuppression is a mainstay of solid organ transplantation and treatment of autoimmune diseases, gene therapy researchers hope that transient immunosuppression only will be necessary for gene therapy.
A third solution is to use viral capsids from serotypes that do not normally infect humans. Even though AAV-2 is non-pathogenic in humans, most humans have pre-existing immunity to this serotype. One solution is to use AAV from serotypes for which humans do not have pre-existing immunity. In this way, one might be able to avoid the immediate clearance of virally transduced cells. Using alternative serotypes, however, will not avoid the problem of elimination of transduced cells by the adaptive immune response.
Protecting the Genome
Cancer biologists have used the insertional activation of proto-oncogenes by retroviral insertion to transform cells for decades. The French SCID-Xl and German CGD trial showed that insertional oncogenesis could also occur from gene therapy using replication incompetent retroviral vectors. One of the areas of future research to determine how to protect the genome from the effects of retroviral insertion and thereby minimize or even prevent insertional oncogenesis.
Improving Viral Vectors
In the retroviral trials that resulted in leukemia or myeloproliferation the transgene was expressed through the LTR. The viral LTR also had the unfortunate effect of activating neighboring proto-oncogenes. One solution is to use self-inactivating (SIN) retroviruses.(17) In SIN viruses, the LTR is modified so that when the retrovirus integrates into the genome the promoter/enhancer activity is inactivated. The expression of the transgene would then be driven by an internal promoter, which would be designed to have a lower probability of activating neighboring genes. Pre-clinical studies will have to determine the magnitude to which SIN viruses do not activate neighboring genes in comparison with non-SIN viruses.
Insulators
One potential way to protect the genome from the effects of retroviral insertion (and the transgene from position effect variegation) is the use of insulators. Insulators are small DNA elements that act as barriers between different segments of the genome.(18) When an insulator is placed between a promoter/enhancer and a gene, the promoter/enhancer can no longer effect the expression of that gene. In addition, if an insulator is placed between heterochromatin, regions of DNA that are silent and tend to spread and silence the expression of nearby genes, it also prevents heterochromatic spreading of silencing. Several groups of investigators are studying how well insulators can buffer the genome and the viral integrations from each other. Encouraging data has emerged that position effect variegation is minimized by incorporating insulators into gene therapy vectors.(19),(20) By minimizing position effect variegation, each viral integration is less likely to be silenced, more likely to create the protein of interest and thus more likely to result in clinical benefit. Studies are now ongoing to determine how well insulators protect the surrounding genome (the prevention of insertional oncogenesis) from the effects of promoter/enhancer elements within the gene therapy vector.
Targeting Integration
Another solution to the problem of insertional oncogenesis is to design ways to target the transgene insertion to a specific location in the genome. That is, instead of letting the retrovirus or lentivirus integrate into the regulatory regions or coding regions of random genes as they are biased to do, researchers could target transgene integration to a specific genomic location that has been prospectively identified to be both safe and permissive -- safe because if the transgene integrates there it will not activate a proto-oncogene or inactivate a tumor suppressor and permissive because if the transgene integrates there it will express the transgene at therapeutic levels.
There are several possible ways to target transgene integration. The most established is the strategy developed by Michelle Calos at Stanford University. Her group has done extensive pre-clinical testing of the phiC31 phage integrase system. The phiC31 integrase, derived from Streptomyces, catalyzes recombination between attB and attP sites. In this way, the enzyme can take a piece of DNA that has an attB site and insert it into a piece of DNA that has an attP site. Calos and colleagues have shown that if they co-introduce the phiC31 integrase protein, along with the transgene that contains an attB site, the integrase will catalyze the integration of the transgene into the genome at an attP site. In pre-clinical testing, they have shown that efficacy in models of tyrosinemia type I, epidermoyisis bullosa, factor IX deficiency and SCID-Xl.(21),(22),(23),(24)
One of the current safety issues in the use of phiC31-mediated integration is whether it induces genomic instability. Studies of the human genome have shown that there are thousands of potential attP sites, thus the specificity of integration may not be as precise as one would hope. Moreover, there is also concern that the phiC31 integrase may create genomic instability by causing translocations induced by recombination between occult attB and attP sites in the genome. Studies to assess the degree of genomic instability induced by phiC31 and ways to improve the specificity of phiC31 are ongoing.(25)
Using Homologous Recombination to Correct Mutations
The most precise way to manipulate the genome of any cell is by gene targeting via homologous recombination. In gene targeting, an introduced DNA fragment replaces a homologous endogenous gene fragment (see Figure 1). In this way, the sequence changes in the introduced DNA fragment can be precisely introduced into the endogenous gene, while leaving the rest of the genome unperturbed. This technique is widely employed experimentally and has been used to manipulate the genomes of bacteria, yeast, chicken DT40 cells and murine embryonic stem (ES) cells with profound scientific impact. For instance, by using gene targeting in murine ES cells, one can create ES lines with experimentally defined changes in specific genes. These ES cells can then be used to generate genetically modified mice. In this way, mouse models of thousands of human diseases have been created. The precision of gene targeting by homologous recombination makes it a potentially ideal way to correct genetic mutations that cause human disease.
When one introduces a DNA fragment into human somatic cells and determines the rate of gene targeting, one finds that only one cell out of every million will undergo gene targeting.(26) This extremely low spontaneous rate of gene targeting has discouraged investigators from exploring homologous recombination as a tool for gene correction gene therapy. In the last several years, however, two different approaches to stimulating the rate of gene targeting in human somatic cells have been discovered:
- the use rAAV vectors;
- the use of DNA double-strand breaks.
In 1998, Russell and Hirata reported that cells infected with rAAV underwent a high rate of homologous recombination(27) and could correct a variety of lesions in a wide range of cell types.(28) Building on this work, the Russell group then used rAAV-mediated homologous recombination in mesenchymal stem cells to inactivate a dominant mutation in the collagen Col1A1 gene that causes osteogenesis imperfecta.(29) By inactivating the dominant mutation, they demonstrated that the cells had phenotypic correction of the underlying defect in this mouse model. Several issues remain with rAAV-mediated homologous recombination. To obtain recombination rates of useful magnitude, infection of cells with high multiplicity of infections (MOIs) is required, increasing the incidence of random integration. In fact, most integration events will be non-homologous rather than homologous. A study of these non-homologous rAAV integration events showed that many were associated with chromosomal rearrangements and occurred in the control regions of genes.(30),(31)
A second method of stimulating homologous recombination is by inducing DNA double-strand breaks (DSBs) in the target locus. In the mid 1990s, a number of labs demonstrated that a specific DSB in a genomic target created by the I-SceI homing endonuclease stimulated homologous recombination between the genomic target and transfected plasmid ("gene targeting") by a thousand fold.(32) With optimization, gene targeting rates of 3-5% can be obtained.(26) The limitation of this approach is that endogenous genes do not contain recognition sites for I-SceI.
If DSBs are to be used to stimulate gene targeting, a method to create site-specific DSBs needed to be developed. Zinc finger nucleases (ZFNs) have shown promise in being such a reagent. These nucleases are artificial proteins in which a zinc finger DNA binding domain is fused to the nuclease domain derived from the type IIS restriction enzyme FokI and were first developed by Chandrasegaran and his colleagues.(33),(34) Zinc finger DNA binding domains are 30-amino acid polypeptides that bind a single Zn++ molecule, fold into ββα configuration and bind to DNA. A single zinc finger domain binds to a triplet of nucleotides. Zinc finger domains can be strung together in series. When three zinc finger domains are linked together, the protein will bind a specific nine-base pair sequence. By linking the nuclease domain to the zinc finger domain, the nuclease now cuts DNA at a specific DNA sequence (determined by the zinc finger portion) instead of randomly.
The first demonstration that ZFNs might be useful for gene therapy was in 2003 when Porteus and Baltimore showed that model ZFNs could stimulate gene targeting by several-thousand fold.(26) Urnov et al. (2005) then demonstrated that ZFNs could be designed to recognize an endogenous target gene, in this case the IL2RG gene, and create targeting rates of 20% in the human K562 cell line and 5% in primary human T-cells.(35) These results inspire optimism about the possibility of using ZFNs to stimulate homologous recombination for therapeutic purposes.
One of the appeals of ZFNs is that they can be modified to recognize novel target sequences. Theoretically it may be possible to make ZFNs essentially directed at any target sequence but a current area of active research is to establish the best practical method to do this. Barbas and his colleagues have released a program that allows users to examine a sequence of interest for binding sites to which zinc fingers can be assembled (www.zincfingertools.org). Whether this relatively simple modular-assembly approach is good enough to make highly active and specific ZFNs or whether more complex selection based approaches are needed remains to be determined.
In the continued development of ZFNs, it is important to demonstrate that ZFNs do not create oncogenic mutations. DSBs are known to be mutagenic and can lead to chromosomal translocations. Studies of ZFNs in model organisms and mammalian cells have demonstrated that they cause cytotoxicity by the creation of DSBs.(26),(36) It is critical, therefore, to design ZFN systems that minimize these off-target DSBs and to show that these systems do not cause oncogenic mutations/translocations using the cell-based and mouse model systems described earlier. An additional potential problem is that the donor DNA can also integrate in a random fashion. While the donor can be engineered so it is promoter- and enhancer-less to minimize insertional activation of nearby genes, the random integration of the donor could still potentially inactivate genes (such as tumor suppressors).
We have discussed using rAAV and DSBs as independent ways to stimulate homologous recombination. Two papers have shown that rAAV and DSBs generated using the I-SceI endonuclease can act together to stimulate homologous recombination.(37),(38) Studies to determine if rAAV and ZFNs act in a similar manner are ongoing.
Using Stem Cells for Gene Therapy
For gene therapy to have long-term efficacy, it must occur at some level in stem cells. Stem cells, by definition, are cells that can undergo self-renewal and can repopulate the mature, differentiated cells of a tissue or organ. The best-characterized stem cell for medical purposes is the hematopoietic stem cell (HSC). The HSCs can be removed from the body, identified by molecular markers, manipulated in vitro for a short period of time, stored for long periods of time and infused into a patient where they can re-constitute the entire hematopoietic system. For monogenic diseases of the hematopoietic system, such as sickle cell disease, David Baltimore in 1978 elucidated the still current working paradigm:(39)
...it would be a triumph of medicine if the effects of such genes could be countered... One approach involves altering some cells of the body so that they can carry out the needed function. A patient could, for instance, be treated in this way for a blood disease caused by an abnormal protein made by a mutant gene. A normal gene would be inserted into the precursor cells' immature bone marrow cells that ultimately develop into functioning blood cells. In this way, a normal protein could be made in place of, or along with, the aberrant protein. The genetically altered blood cell precursor could then cure the patient's disease...
One of the major limitations of this paradigm is generating enough HSCs that have undergone gene therapy to be of therapeutic benefit and to show that collection of HSCs does not contain a rare cell that has acquired a pre-disposing oncogenic change in the process. One solution would be if we could expand HSCs in vitro. In this way, a small population of HSCs that were prospectively identified to be both safe and efficacious could be identified and subsequently expanded to sufficient numbers to reconstitute the hematopoietic system after re-infusion. Unfortunately, the ability to expand human HSCs in vitro does not exist. Since HSCs undergo such expansion in vivo, it is assumed that if the right conditions are found, such expansion could be performed in vitro. Current research looking at such molecules as HoxB4, Wnt and Notch, and the interaction between the HSC and its stem cell "niche" are aimed at developing such protocol.
An alternative approach is to use human embryonic stem cells to generate enough tissue-specific stem cells. Embryonic stem (ES) cells are derived from the inner cell mass of a blastocyst. Once an ES line is derived it can expand indefinitely without acquiring oncogenic mutations and can differentiate into a wide variety of cell types, such as neurons, blood cells, fat, muscle. One strategy, therefore, is to perform gene therapy in patient-specific embryonic stem cells, characterize those cells to show that they are both safe and efficacious, expand the manipulated ES cells to sufficient quantities in vitro, and then infuse those cells into the patient for therapeutic purposes. Since the infusion of undifferentiated ES cells results in the generation of teratocarcinomas, the undifferentiated ES cells must first be differentiated into tissue-specific stem cells, such as hematopoietic stem cells or neuronal stem cells, before infusing. There are a number of researchers who are actively exploring how to differentiate reliably and consistently human ES cells into tissue-specific stem cells so that they can be used for therapeutic purposes.(40) These studies are very much in their infancy and the ability to perform gene therapy in this way remains at least a decade away.
The development of leukemia in the French SCID-Xl trial and myeloproliferation in the German CGD trial highlight that we need to develop better pre-clinical models for the safety of gene therapy. In both of these trials, pre-clinical testing of efficacy had been done by studying mouse models of either SCID-Xl or CGD. In retrospect, these pre-clinical studies were not designed properly to be good pre-clinical tests of safety. Subsequent studies in mice, for example, have shown that the leukemias might have been predicted if the pre-clinical animal models had been done differently.(41),(42) Woods et al. (2006) showed that 33% of mice infused with bone marrow cells transduced with a lentiviral vector expressing the IL2RG gene developed leukemia but that the leukemias took over six months to appear.(42) In the pre-clinical studies of efficacy for the SCID-Xl trial, the mice were all sacrificed before six months of age and so the study was not structured to detect the serious adverse events that subsequently occurred in the human trial. In the future, quantitative models of safety, both cell- and animal-based, need to be developed to assess each gene therapy protocol. Once these models are developed and validated, more rational decisions about the risks and benefits of gene therapy can be made. Moreover, as gene therapy vectors and protocols evolve, these quantitative models will allow us to quantitate how much, if any, these changes improve the safety of the procedure.
Ethics of Gene Therapy
Given the perceived lack of success of gene therapy, it might seem like putting the cart before the horse in terms of talking about the ethics of gene therapy. Like most technologies, however, once gene therapy is generally successful, as I believe it will one day be, it is better if the ethical groundwork had been laid. For gene therapy, one can conceive of a number of important ethical questions. Here I will focus on three:
- Should we be manipulating the genome of human beings at all?
- How do we choose the correct patients for gene therapy trials?
- How do we prevent gene therapy from being used for non-medical purposes?
When recombinant DNA was first developed, an international debate ensued about whether scientists should even use these new techniques. There was concern that the ability to splice together pieces of DNA that were not naturally found together would create chimeras, abnormal life forms that either would be directly harmful to human society or were more morally reprehensible and should not be made. There was concern that recombinant DNA allowed humans to create things that were beyond their purview. With an open, although not always cordial, debate among the various interested parties came a consensus that recombinant DNA technology, with appropriate regulation, was worth using. The subsequent medical benefits from that consensus are too numerous to enumerate.
But should we be using recombinant DNA technology to alter the human genome? The answer to this question depends on which cells we are altering. If we are discussing human somatic cells, cells that have no ability to become germ cells, then there is a clear consensus that it is ethically reasonable to pursue such a strategy because it may result in the improved health of individuals without altering the human genetic code that is passed from generation to generation. On the other hand, I personally believe that even if using gene therapy to manipulate the genome of germ cells was possible it should not be done. The consequences of such manipulation are currently beyond the scope of our understanding. For that reason, gene transfer into human germ cells is a line that should not be crossed, either intentionally or unintentionally.
There are wise physicians, scientists, philosophers and ethicists, however, who believe that gene therapy of germ line cells is not only ethically permissible but ultimately ethically necessary, as it may be the only way to cure certain monogenic diseases, such as Li-Fraumeni syndrome (a cancer predisposition syndrome caused by mutations in the p53 gene) or cystic fibrosis (a multi-system disease caused by mutations in the CFTR gene). In our current political climate, however, it seems unlikely that an open and honest debate about these issues can be had.
The second ethical issue is which patients should be enlisted to participate in experimental gene therapy trials? In this respect, gene therapy is not fundamentally different from other clinical trials of newly developing technologies or therapies. Our current system of institutional review boards, while not without its flaws, is probably capable of handling the ethical issues of patient selection for gene therapy trials. The decision to exclude severely affected patients from the OTC trial for ethical reasons is one that was carefully considered but I believe that an ethically permissible structure can be found to allow patients who have no other options to participate in gene therapy trials. In fact, one of the important factors in assessing the ethics of a gene therapy trial, like any clinical trial, is balancing the risks and potential benefits of the experimental therapy with the quality of life gained from currently available therapies. There is no doubt that people and patients will have differing views on where this balance should lie based on a multitude of individual, familial, social, religious and cultural factors. An ethical gene therapy trial, like any trial of a new therapy, must allow these varying considerations to be accounted for. Moreover, doctors and investigators must recognize the powerful position they hold when they offer a seeming magic bullet to patients or parents who often perceive that they have limited sub-optimal options.
Finally, what about the ethics of using gene therapy for non-medical purposes such as for personal enhancement like cosmetic surgery or for performance enhancement, as athletes use anabolic steroids, human growth hormone or erythropoietin? The fact that gene therapy is not completely safe is not necessarily going to dissuade people from using it. The adverse health effects of continued use of anabolic steroids are well publicized, yet many (perhaps millions) people use them or have used them to enhance their athletic performance. Yet, I think there would be a consensus that it is ethically inappropriate to modify our genomic DNA by gene therapy for personal or performance enhancement. Such uses need to be anticipated, creative thinking applied to the potential ways gene therapy could be misused and regulatory frameworks established to prevent the misuse of gene therapy.
Summary
Medical history is littered with examples, such as solid organ and bone marrow transplantation, of how profoundly important therapies are characterized by fits and starts in their development. When gene therapy for monogenic diseases was first conceived in the 1970s after the development of recombinant DNA technology, it seemed that it might not have to go through the usual growing pains. The last thirty years, however, has shown that gene therapy is not immune from these developmental hiccups. As the field has matured, it is now recognized that gene therapy will become part of standard medical therapy, not by hitting a home-run, but instead by incremental and sustained advances ("Billy-ball" for those fans of the Oakland A's in the pre-steroid era), just as pediatric acute lymphoblastic leukemia was transformed from a universally fatal disease to one that has now has cure rate of 90%.
Notwithstanding the high-profile adverse events for gene therapy clinical trials, there are now an increasing number of patients whose lives have been improved by gene therapy. Moreover, there are a number of exciting emerging avenues of future research, the use of homologous recombination, the development of insulators, the continued improvement of viral vectors and the great potential of stem cells, that bode well that gene therapy will eventually develop into a mainstay of medical therapy.