Course Authors
Nicole A. Shonka, M.D., and Mark R. Gilbert, M.D.
Release Date: 02/22/2010
Upon completion of this Cyberounds®, you should be able to:
Highlight pathways important in gliomagenesis
Discuss the current use of molecularly targeted agents for gliomas, as well as efficacy and adverse effects of treatment
Discuss the ongoing research using targeted agents in the treatment of gliomas.
Gliomas are the most frequently occurring malignant primary brain tumors and include astrocytomas, oligodendrogliomas and ependymomas. Combined, these histologies account for approximately 40% of all primary brain tumors and over 80% of all malignant CNS tumors.(1) Typically, high−grade gliomas (Grades III and IV) are referred to as “malignant” glioma. As glioblastoma (GBM), the only grade IV glioma, accounts for approximately 50% of gliomas,(1) a majority of glioma research has focused on this tumor type (see Figure 1).

Figure 1. Central Brain Tumor Registry of the United States: Primary Brain Tumors By Histology.

The current standard therapy for GBM changed in the last five years. The report of a large prospective randomized phase III trial from the European Organization for Research and Treatment of Cancer (EORTC) and The National Cancer Institute (NCI) of Canada supported the use of concurrent chemotherapy with temozolomide, with standard conformal radiotherapy followed by adjuvant temozolomide. This trial randomized 573 patients from 85 centers to receive either standard radiotherapy or concurrent temozolomide with radiotherapy followed by adjuvant temozolomide for 6 months. The group receiving concurrent and adjuvant temozolomide had a significant improvement in progression free survival (7.2 versus 5.0 months), median survival (15 versus 12 months), and 2−year survival rate (26% versus 8%).(2)
Almost inevitably, however, patients will progress after traditional chemotherapy. The field of neuro−oncology is focusing on the uses of molecularly targeted agents in the adjuvant setting. This Cyberounds® CME activity aims to highlight the pathways important in gliomagenesis and to detail the mechanisms by which targeted therapy may act upon tumor cells.
The Molecular Biology of Gliomas
Three major molecular pathways have been identified as key in the development of gliomas (see Figure 2).(3) First, either an extracellular growth factor activates a receptor tyrosine kinase (RTK) such as epidermal growth factor receptor (EGFR) or platelet−derived growth factor receptor (PDGFR), or an activating mutation renders the receptor constitutively active. The pathway marked ‘A’ illustrates the signal transduction via RTK through the AKT signaling pathway by activating PI3K. Pathway ‘B’ can be activated simultaneously or alone and transduces its signal through the RAS− RAF−MEK−ERK cascade. Pathway ‘C’ depicts CDK4 (INK4A)−ARF which encodes two gene products that inhibit cell division. In the upper arm, INK4A inhibits the activity of CDK, leaving RB free to bind to and inactivate E2F. In the lower arm, ARF forms an inhibitor complex with MDM2, to prevent its inhibition of p53 (which functions as a tumor suppressor).(3)
Figure 2. Molecular Pathways In Glioma.

Click image to view large scale.
Further understanding of gliomagenesis has permitted the evaluation of targeted agents to inhibit specific signaling pathways. Targets have included growth factor receptors such as PDGFR and EGFR, Raf and the mammalian target of rapamycin (mTOR). Other targets have included the vascular endothelial growth factor and its receptor (VEGF and VEGFR) as well as intrinsic components of DNA structure (see Figure 3). We will highlight those at the forefront in translational research and those already used in practice.
Figure 3. Glioma Signaling Targets.
Click image to view large scale.
Angiogenesis Inhibitors
As our understanding of gliomagenesis grows, so do the number of potential targets at which to aim therapy. Angiogenesis has been identified as a prime therapeutic target because glioblastoma is among the most vascularized tumors. Glioblastoma obtains its vasculature via: (1) co−option of existing vessels, (2) classical angiogenesis (inducing capillary growth from pre−existing vessels through proliferation and migration of endothelial cells) and (3) vasculogenesis (attraction of bone marrow−derived cells which stimulate vessel growth out of the blood and into the perivascular space).(4)(5)(6) Vascular endothelial growth factor (VEGF) induces tumor angiogenesis, promotes edema by increasing vascular permeablility and promotes vasculogenesis.(6)
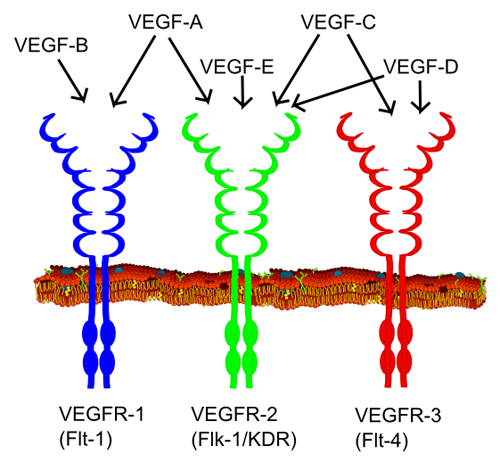
All members of the VEGF family bind to VEGF receptors (VEGFR) which are tyrosine kinase receptors found on the cell surface. This leads to dimerization and activation via phosphorylation. VEGFRs are composed of seven extracellular immunoglobulin−like domains, a region that spans the cell membrane, and an intracellular region of a tyrosine kinase domain.(7) VEGF−A binds to both VEGFR−1 and VEGFR−2. VEGFR−2 plays a central role in most cellular responses to VEGF, while the role of VEGFR−1 is thought primarily to modulate signaling of VEGFR−2, and to sequester VEGF away from VEGFR−2. VEGFR−3 mediates lymphangiogenesis in response to VEGF−C and VEGF−D (see Figure 4).(7) Figure 5 depicts the VEGF pathway.
Figure 4. VEGF Family and Receptors. Source: Wikipedia
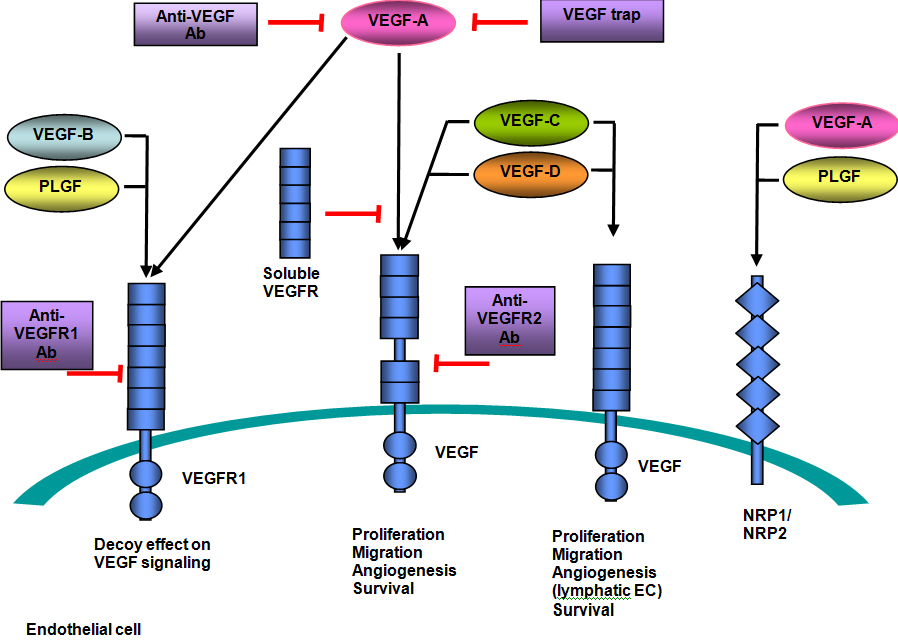
Bevacizumab
Bevacizumab, as the nomenclature suggests, is a monoclonal antibody directed against all isoforms of VEGF−A. It binds to VEGF to prevent its interaction with surface VEGFRs to inhibit VEGFR signaling. Most recently, bevacizumab has been approved by the FDA for recurrent GBM following first−line therapy based on two clinical trials. One study showed a 6−month progression free survival (PFS) rate of 42% and overall survival (OS) of 8.7 months in patients receiving bevacizumab alone.(8) Another study showed a 19.6% objective response rate with median duration of 3.9 months. Six−month PFS was 29% and six−month survival was 57%. Additionally, 50% of patients experienced decreased cerebral edema, 58% were able to decrease their corticosteroid dependency and 52% had improvement in their neurologic symptoms.(9) Adverse effects include wound healing delays, bleeding complications, arterial thromboembolic events, proteinuria and hypertension in up to 30% of patients. Despite its humanized antibody form, up to 5% of patients can have infusion−related symptoms of fever, chills, urticaria, bronchospasm, angioedema and hypotension.(10)
Cediranib
Cediranib is a pan−VEGFR tyrosine kinase inhibitor with activity against PDGFR as well as c−Kit. Evaluated in 16 patients with recurrent glioblastoma, it demonstrated an ability to decrease tumor−associated vasogenic brain edema.(11) In a phase I study with cediranib, fatigue, diarrhea, nausea and anorexia were commonly noted. Grade 3 or 4 hypertension occurred in almost 20% of patients.(12)
However, despite the results reported above, it has not been established that VEGF inhibitors truly have anti−tumor effects or if the imaging improvements seen are from decreasing permeability effects with a resultant decrease in tumor enhancement and edema. One trial utilized orthotopic xenografts (i.e., transplanting live cells from one species to another) of GBM in mice and found that cediranib resulted in a significant increase in OS despite persistent tumor growth(13). For this reason, it has been postulated that agents directed against VEGF may best be used in combination with cytotoxic agents. Many studies are ongoing, including evaluating bevacizumab given concurrently with temozolomide and radiation for primary GBM, as well utilizing cediranib in combination with lomustine for recurrent GBM.
Figure 5. VEGF Pathway.
Click image to view large scale.
Growth Factor Receptors
EGFR
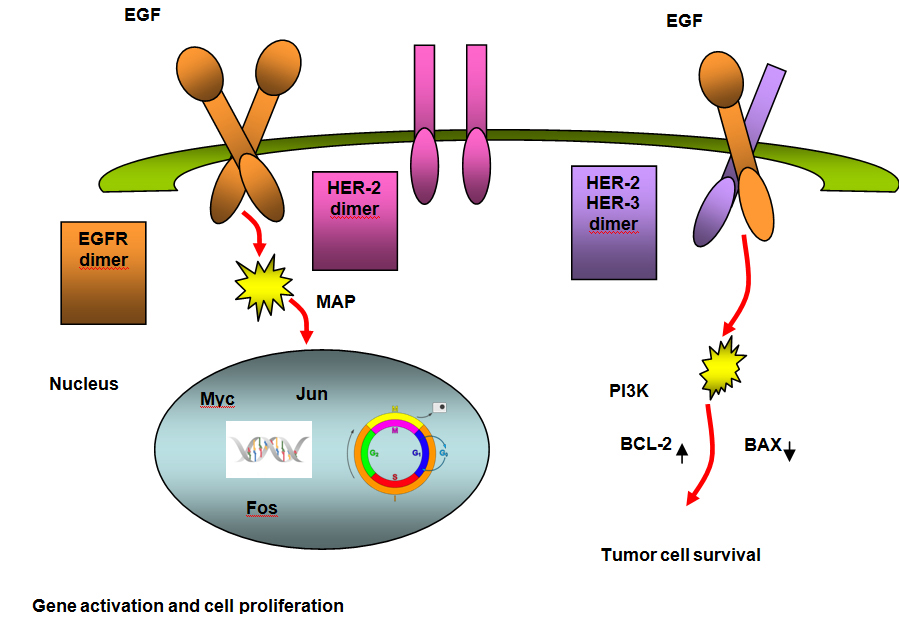
The epidermal growth factor receptor (EGFR) is the first member of a structurally similar group of receptors known as the erbB receptor family. There are four members in this family, including EGFR/HER−1 (c−erbB1), HER−2 (c−erbB2), HER−3 (c−erbB3) and HER−4 (c−erbB4). These receptors function as heterodimers if more than one receptor type is present. Growth factor− receptor binding causes dimerization and autophosphorylation on tyrosine residues (see Figure 6 parts A and B).(14)
Figure 6A. EGFR: Normal Signaling.
Click image to view large scale.
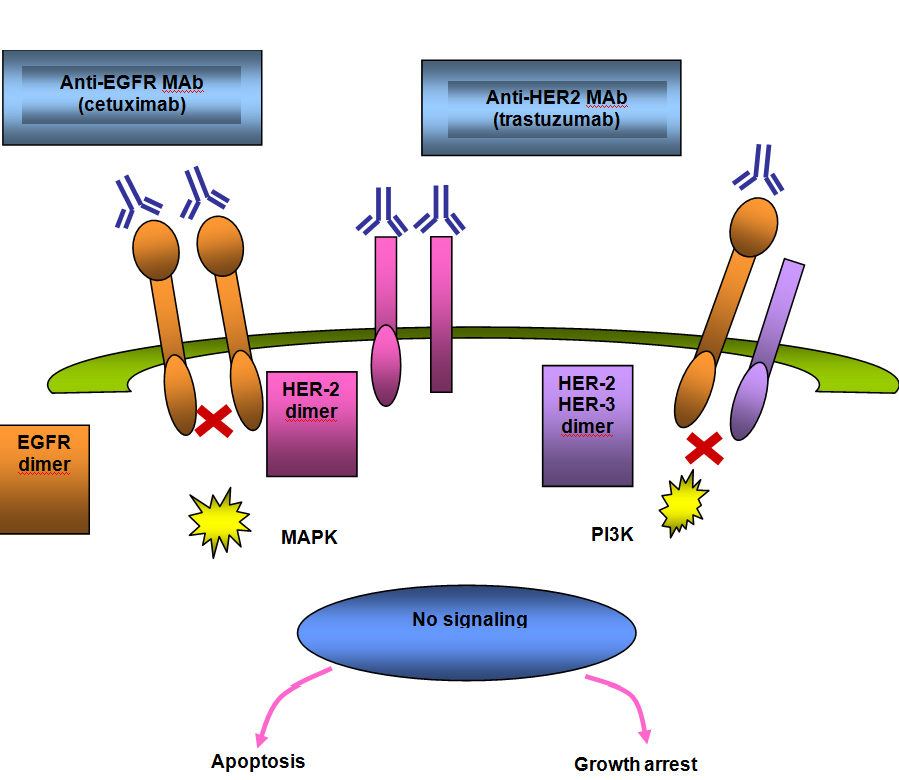
Figure 6B. Signal Blocked By Anti−EGFR Agent.
Click image to view large scale.
The gene encoding EGFR is located on chromosome 7 and is the most frequently amplified gene in GBM,(15) occurring in approximately 40% of primary GBM.(16) EGFR amplification occurs only rarely in secondary GBM and in one study was not detected in any patients under the age of 35 years.(16) The most common variant in GBM is EGFRvIII, present in up to 50% of GBM that have amplification of EGFR. EGFRvIII is tumor−specific and found on the cell surface, making it an attractive target for therapy.(17)(18) The EGFRvIII results in constituative activation of the pathway and leads to proliferation through the PI3−kinase and RAS pathways.(19) EGFR can be blocked using small−molecule tyrosine kinase inhibitors which interfere with ATP−receptor binding, or by monoclonal antibodies which bind to the extracellular portion of the receptor, thereby inhibiting dimerization and autophosphorylation.
Erlotinib
Erlotinib is a signal transduction inhibitor targeted against the EGFR tyrosine kinase. One−hundred and ten patients with GBM recurrent after radiation therapy (RT) were randomly assigned to either single agent erlotinib or cytotoxic chemotherapy with either temozolomide or carmustine. Pathology on tumor tissue at time of diagnosis was evaluated for EGFR expression, EGFRvIII, EGFR amplification and EGFR mutations in exons 18, 19 and 21. Skin toxicity was the most frequent adverse effect of erlotinib. The 6−month PFS rate in the erlotinib arm was 11.4% compared with 24% in the control arm, and no biomarker was found to correlate with increased efficacy. (20) The most common adverse effects include a pustular, acneiform rash that can be quite disfiguring (see Image 1), diarrhea, anorexia, mild elevations in transaminases and rarely pulmonary toxicity.(10)
Image 1. Erlotinib Rash.
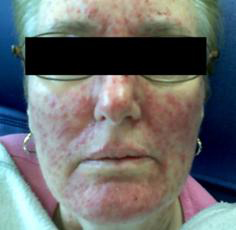
Source: JOP. Saif, W. J Pancreas (Online) 2009 May 18; 10(3):338−340. Used with permission.
Nimotuzumab and Cetuximab
Since the correlation between EGFR expression and radioresistance was noted in head and neck cancer,(21) manipulating this target has become of interest to increase radiosensitivity. Cetuximab is a chimeric antibody that binds to domain III and blocks EGF binding by sterically interfering with the active conformation. Nimotuzumab is a humanized antibody which also binds to domain III of the extracellular region of the EGFR but in a location toward the c−terminus of that domain, which allows domain I to approach domain III to result in active conformation. This allows a basal level of signaling to occur, which likely explains the lack of adverse effects seen with nimotuzumab in comparison with other agents blocking EGFR, specifically rash.(22)
Mice inoculated with a human GBM xenograft (U87MG) were randomized to nimotuzumab or cetuximab either with or without radiation, and compared to those given radiation alone. The addition of either antibody to radiation resulted in improved response and a reduced number of radioresistant cancer stem cells; nimotuzumab also resulted in a reduction in tumor vasculature.(23) A phase III study using nimotuzumab in addition to temozolomide and RT for treatment of GBM is enrolling currently in Germany.
PDGFR
In the platelet derived growth factor (PDGF) family there are five isoforms of PDGF: PDGFA, PDGFB, PDGFC, PDGFD and an AB heterodimer. These ligands act upon two different receptors (PDGFR): alpha (PDGFR−alpha) and beta (PDGFR−beta). As with VEGF and EGF signaling, PDGF also works by activating the tyrosine kinase receptors (in this case PDGFR) found on the cell surface, leading to dimerization and activation of intracellular pathways via phosphorylation.(24)
PDGFR mutations and amplification are rare, but overexpression of PDGFA and/or PDGFB, as well as their receptors, is found in diffuse glioma. Moreover, the co−expression of the ligand−receptor pairs has been shown to demonstrate autocrine activity. Levels of PDGF and PDGFR increase in higher grade gliomas. Notably, almost all oligodendrogliomas express PDGF and PDGFR.(25) Blocking PDGF signal transduction has been shown to inhibit growth in glioma cell lines.(26)
Imatinib
Imatinib is a signal transduction inhibitor that is well known for its inhibition of the BCR−ABL tyrosine kinase in chronic myelogenous leukemia, and it also inhibits other tyrosine kinases including PDGFR, stem cell factor and c−kit. The most common toxicities from imatinib therapy are nausea and vomiting which are seen in approximately 40% of patients, followed by diarrhea in 30%. Other adverse effects include pedal and periorbital edema. Fluid retention with pleural effusions and ascites can be seen more often in the elderly and at higher doses. Cardiac function should be regularly monitored with multiple gated acquisition scan (MUGA) or echocardiogram at baseline and periodically. Myelosuppression can be seen, particularly neutropenia and thrombocytopenia.(10)
In a phase II study, 112 patients with recurrent glioma [51 patients with GBM, 25 patients with anaplastic astrocytoma (AA) and 36 patients with oligodendroglioma (OD) were given single−agent imatinib. No somatic activating mutations of c−kit or PDGFR−alpha or −beta were found. Five patients had a partial response (PR), including three patients with GBM, and the 6−month PFS (PFS−6) was 16% in GBM, 9% in AA, and 4.0% in OD.(27) A PFS−6 of less than 15% in recurrent glioma is generally accepted as indicating an inactive regimen, so the PFS found in this trial may point to a low level of activity of imatinib for GBM. This is in contrast to a PFS−6 in GBM of only 3% and PFS−6 of 10% in AA found in a phase I/II trial of imatinib in 35 patients.(28) After promising data from a phase II trial, imatinib was given with hydroxyurea to patients with recurrent GBM in a phase III trial randomizing patients to either single agent hydroxyurea or combined with imatinib. However, in this large multicenter trial, the PFS−6 was only 5% in the combination arm and 7% in the monotherapy arm.(29)
Growth Factor Effectors
mTOR
As shown in Figure 3, the phosphatidylinositol−3−kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway plays a crucial role in regulating glioma cell growth, survival and proliferation. Loss of PTEN, common in GBM, results in the activation of mTOR.(30) This subsequent activation of the PI3K/Akt/mTOR pathway has been associated with poor prognosis in AA, anaplastic oligodendroglioma (AOD) and GBM.(31) The immunosuppressant sirolimus (rapamycin) directly inhibits mTOR activity and has been shown to hinder the growth of cancer cells in vitro and in vivo. As a consequence of this finding, sirolimus derivatives have been developed as targeted therapies.
Temsirolimus, an ester of sirolimus, causes cell−cycle arrest at G1 by inhibiting signaling pathways that result in the inhibition of RNA translation. Its adverse effect profile includes myelosuppression, nausea, vomiting and diarrhea, hypertriglyceridemia and cutaneous toxicity.(32) Two phase II studies of temsirolimus in recurrent GBM have been reported: in the first, despite improved imaging in 36% of patients, PFS−6 was only 7.8%, (33) and in the second only one of 41 patients was free from progression at six months.(34) A number of studies are ongoing both in the newly diagnosed and recurrent GBM settings, with temsirolimus added to RT, temozolomide, sorafenib, erlotinib, tipifarnib and bevacizumab.
PKC−beta Inhibitors
Enzastaurin
Activation of protein kinase C−beta (PKC−beta), implicated in tumor−induced angiogenesis, tumor cell proliferation, apoptosis and invasiveness, is inhibited selectively by enzastaurin. Enzastaurin prevents substrate phosphorylation by competing with ATP for the ATP binding site, which inhibits not only PKC activation but also its target proteins such as AKT (see Figure 7). Enzastaurin treatment suppresses the phosphorylation of AKT, just upstream of mTOR (pathways seen in Figs. 2 and 3).
Figure 7. MAPK Activation via PKC.

Click image to view large scale.
After demonstrating activity in human GBM xenografts,(35) a phase I/II study evaluating enzastaurin as a single agent showed promising efficacy but with the concern that drug levels in the brain were suboptimal.(36) Another phase I study was done at higher doses with GBM and AA patients with recurrent disease, and of 19 evaluable patients, one had a complete response (CR) (shown in Image 2), one a PR, and 8 had stable disease (SD). The median PFS was only 1.4 months, however, and higher doses were poorly tolerated with significant thrombocytopenia and prolonged QTc intervals.(37) Further trials using this agent in combination with cytotoxic chemotherapy or alone in the newly diagnosed and adjuvant settings are ongoing.
HDAC Inhibitors
In recent years, histone deacetylase inhibitors (HDACIs) have emerged as a promising new category of anti−cancer agents. Epigenetic alteration of chromatin functions in nuclear homeostasis as well as in gene expression. Nucleosomes are the basic repeating units that constitute eukaryotic chromatin. Each nucleosome is composed of an octamer of four pairs of core histones (H2A, H2B, H3 and H4) wrapped in DNA. The H3 and H4 histones have long tails which can be modified at several places and the core of histones H2A and H3 can also be modified. Modification of the amino acid tails of histones occur through acetylation, phosphorylation, methylation and ubiquitination.(38) Chromatin acetylation is regulated by the balanced action between histone acetyltransferases (HATs) and histone deacetylases (HDACs). The HATs transfer acetyl groups from acetyl coenzyme A onto the lysine residues within the core histones, neutralizing their positive charges and opening the tightly wound chromatin to allow transcription to take place.(39) HDACIs upset this balance and initiate growth arrest, terminal differentiation and apoptosis (see Figure 6).(38) Although HDACIs cause cell cycle arrest, differentiation and apoptosis in cancer cells, they have little effect on normal cells, making them less toxic as targeted therapy.(40)
Figure 8. Histone Acetylation and Deacetylation.
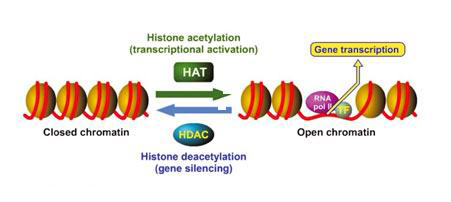
Source: Drugs of the Future 2007; 32(1): 45−50. Copyright © 2007 Prous Science, S.A. All rights reserved. Used with permission.
Vorinostat
Vorinostat inhibits HDACs leading to an accumulation of acetylated histones that alter gene transcription and expression. Vorinostat also acetylates non−histone proteins including many that regulate cell proliferation, stabilization and death such as Rb, Hsp90 and Bcl−2, as well as proteins acting to enhance motility, angiogenesis and protein expression (tubulin, HIF−1alpha, Trx) (see Figure 9).(41) Although the package insert warns that vorinostat can cause prolongation of the QT interval (usually via hypokalemia and hypomagnesemia), which mandates electrolyte and electrocardiograph monitoring, the most commonly seen side effects from a phase II trial included fatigue and thrombocytopenia.(42)
Figure 9. Mechanism of Vorinostat.

Glioma cell lines incubated with vorinostat and then radiated demonstrated a synergistic decrease in clonogenic survival. Not only did the physical modification of chromatin structure amplify cellular damage but changes in oncoproteins Rad51, DNA−PK, EGFR and AKT were also noted. DNA−PK and Rad51 both function in DNA repair and are increased after radiation damage has occurred. Pre−treatment with vorinostat reduced the levels of these repair enzymes. EGFR expression was decreased and AKT activation attenuated to further increase radiosensitivity.(43) Vorinostat was given to 66 patients with recurrent GBM in a phase II trial and although PFS6 was 15.2%, those patients who were free of progression at that endpoint had a median of 11.2 months of disease stability.(42) Although one study with vorinostat alone for recurrent GBM is ongoing, most studies are focusing on the use of vorinostat in combinations with bevacizumab, bortezomib, irinotecan, temozolomide, isotretinoin, carboplatin and radiation.
Other Agents
Thalidomide and Lenalidomide
Basic fibroblast growth factor (b−FGF), in addition to PDGF and VEGF, also helps to promote angiogenesis in malignant gliomas.(44) Thalidomide is believed to exert antiangiogenic effects via both b−FGF as well as VEGF, and inhibits tumor necrosis factor alpha (TNF−alpha). The toxicities of thalidomide, in addition to its well−publicized teratogenicity, include fatigue, peripheral neuropathy, rash and thromboembolism.(10) Lenalidomide, an analog of thalidomide, also inhibits TNF−alpha, b−FGF and VEGF but can also overcome cellular resistance to thalidomide. It shares teratogenicity and thromboembolic complications with thalidomide, has more myelosuppression with neutropenia and thrombocytopenia and only rarely causes sedation and neurologic sequelae.(10)
As a single agent, thalidomide has had only modest activity.(45)(46) In combination therapy, phase II studies of thalidomide in association with bischloronitrosourea (BCNU) or temozolomide have not shown a clear benefit when compared to historical survival.(47)(48) Several trials of thalidomide in combination with chemotherapy (temozolomide, irinotecan, topotecan, procarbazine and carboplatin) and other agents (interferon, celecoxib, isotretinoin) are currently ongoing.
Lenalidomide demonstrated only minimal activity with median time to tumor progression (TTP) under two months and 12.5% PFS−6 in a phase I study of recurrent high−grade glioma patients. The only significant toxicity was a higher risk for thromboembolic disease.(49) Based on the premise that antiangiogenic therapies may increase tumor oxygenation through tumor vasculature normalization, thereby increasing radiosensitivity, a phase I trial of lenalidomide with RT as initial treatment for GBM was done. Time to tumor progression was 5 months and OS was 11 months. Four of 23 patients developed pulmonary emboli despite prophylaxis with aspirin.(50) The rationale for the study proposed that the anti−vascular effects of lenalidomide may be greater when used concurrent with RT. The use of lenalidomide with irinotecan in the recurrent GBM setting is being studied.
Conclusion
Although the utilization of targeted agents in oncology is not new, it is only in the last several years that the discovery of specific molecular pathways in gliomagenesis has allowed neurooncologists to apply these agents to gliomas. Although the elucidation of these pathways allows a more mechanistic approach to treating brain tumors, malignant cells often survive via accessory pathways with alternative signaling. For this reason, multikinase inhibitors such as sorafenib and sunitinib may prove to be more effective, as may combinations of targeted agents discussed here. We hope that in the next decade our understanding of gliomagenesis will continue to evolve and with it a greater appreciation for molecular manipulation that will translate into improved outcomes for our patients.