Course Authors
Bruce S. McEwen, Ph.D.
Dr. McEwen reports no conflict of interest.
This activity is made possible by an unrestricted educational grant from Forest Laboratories.
Estimated course time: 1 hour(s).

Albert Einstein College of Medicine – Montefiore Medical Center designates this enduring material activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
In support of improving patient care, this activity has been planned and implemented by Albert Einstein College of Medicine-Montefiore Medical Center and InterMDnet. Albert Einstein College of Medicine – Montefiore Medical Center is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Upon completion of this Cyberounds®, you should be able to:
Define "allostasis" and "allostatic load" and give examples of each for the metabolic, cardiovascular and immune systems
Describe the role of excitatory amino acids (EAA) in relation to structural remodeling under stress and how this changes as the brain ages
Describe how glucocorticoids and EAA interact in relation to damage produced by ischemic and seizures on brain structures like the hippocampus
Describe why chronic stress may increase or decease vulnerability to insults such as ischemia or seizures.
Every experience we have, whether or not we call it "stressful," elicits a response in the brain and throughout the body that results in some type of adjustment and adaptation to whatever has happened to us. Whether the "event" is getting out of bed in the morning, walking up a flight of stairs, reading the morning newspaper or arguing with our children about getting dressed for school, the brain orchestrates the production of a host of mediators from the neuroendocrine system, the hormones that regulate metabolism, the autonomic nervous system, the immune system and the neurotransmitters, and neuromodulators within the brain itself.
These mediators help us pay attention, learn, remember, mobilize energy, redirect blood flow where it is needed to keep us conscious and alert. They also help us respond when there is a threat -- the "fight or flight" response -- in terms of rapid energy mobilization and also the enhancement of immune function to heal a wound and fight an infection.
We have seen in previous Cyberounds® in this series how the excitatory amino acid neurotransmitters (EAA) and their receptors, representing the most highly expressed neurotransmitter system in the brain, play a key role in learning and memory and in the adaptive plasticity that is associated with acute adaptation to stress, as well as with the structural plasticity that accompanies repeated stress. And we have also seen how the EAA are involved in damage resulting from ischemia and seizures. This apparent paradox -- between protection and adaptation on the one hand and damage on the other hand -- is not unique to the EAA but is a general phenomenon among all the mediators of adaptation.
This Cyberounds® will discuss the paradox of protection and damage in terms of the concepts of allostasis and allostatic load and then return to the topic of how EAA and their receptors, especially NMDA receptors, interact with other mediators during chronic stress, and whether these interactions increase or decrease the risk for permanent damage from additional stress or other insults. A key aspect of the distinction between protection and damage is the time frame of the response -- that is, the difference between well-regulated acute responses that are turned on and shut off efficiently versus the prolonged and dysregulated protection of the same mediators over weeks, months and even years.
Protective and Damaging Effects of the Mediators of Adaptation
Individual differences in the progression of a number of disorders that accumulate with time can be conceptualized as an accumulation of wear and tear of daily experiences, lifestyle and major life stressors which interact with the genetic constitution and predisposing early life experiences.(1),(2),(3) The neurotransmitters of the brain, as well as the neuroendocrine system, autonomic nervous system and immune system, are mediators of adaptation to challenges of daily life, referred to as "allostasis," meaning "maintaining stability through change."(4)
Physiological mediators such as adrenalin from the adrenal medulla, glucocorticoids from the adrenal cortex and cytokines from cells of the immune system act upon receptors in various tissues and organs to produce effects that are adaptive in the short run but can be damaging if the mediators are not shut off when no longer needed. When release of the mediators is not efficiently terminated, their effects on target cells are prolonged, leading to other consequences that may include receptor desensitization and tissue damage. This process has been named "allostatic load,"(5),(6),(7) which refers to the price the tissue or organ pays for an overactive or inefficiently managed allostatic response. Therefore, allostatic load refers to the "cost" of adaptation.
For example, glucocorticoids, so-named because of their ability to promote conversion of protein and lipids to usable carbohydrates, serve the body well in the short run by replenishing energy reserves after a period of activity, like running away from a predator in the "fight or flight" response. Glucocorticoids also act on the brain to increase appetite for food and to increase locomotor activity and food seeking behavior,(8) thus regulating behaviors which control energy intake and expenditure. This is very useful when we have to run two miles, but it is not beneficial when we grab a bag of potato chips while working on income tax returns.
Inactivity and lack of energy expenditure create a situation where chronically elevated glucocorticoids can impede the action of insulin to promote glucose uptake. One of the results of this interaction is that insulin levels increase, and, together, insulin and glucocorticoid elevations promote the deposition of body fat and this combination of hormones also promotes the formation of atherosclerotic plaques in the coronary arteries.(9) Yet, we also now know that regular, aerobic exercise by normally sedentary people (as little as 30' per day over a number of years) reduces the risk for diabetes and improves cognitive function(10),(11),(12).
For the heart, we see a similar paradoxical biphasic role of allostasis mediators. As noted above, getting out of bed in the morning requires an increase in blood pressure and a reapportioning of blood flow to the head so we can stand up and not faint.(4) Blood pressure rises and falls during the day as physical and emotional demands change, providing adequate blood flow as needed. Yet repeatedly elevated blood pressure promotes generation of atherosclerotic plaques, particularly when combined with a supply of cholesterol, lipids and oxygen free radicals that damage the coronary artery walls.(13) Beta adrenergic receptor blockers are known to inhibit this cascade of events and to slow down the atherosclerosis that is accelerated in dominant male cynomologus monkeys exposed to an unstable dominance hierarchy.(14) Thus catecholamines and the combination of glucocorticoids and insulin, along with a calorie-rich diet, can have dangerous effects on the body, besides their important short-term adaptive roles.(9)
For the immune system, the protective and damaging effects of stress mediators are illustrated by opposite effects of acute versus chronic stress on skin immunity: Acute stress in mice and rats has been shown to induce a significant enhancement of skin immunity.(15) A stress-induced trafficking of leukocytes from the blood to the skin is one of the mediators of this immunoenhancement.(16) Importantly, both the stress-induced changes in leukocyte trafficking and the stress-induced enhancement of immune function, measured as delayed-type hypersenstivity or DTH, have been shown to be dependent on adrenal stress hormones.(15),(17)
However, in contrast to acute stress, chronic stress significantly suppresses skin immunity and impairs leukocyte trafficking, and this may be one of the causes of the immunosuppression.(18) Thus, in this case, allostatic load imposed by chronic stress has detrimental consequences for immune function, although it is not clear what mechanism is involved in suppresssing DTH responses. One possibility is that repeated stress induces a down-regulation of immune cell trafficking and of the movement of immune cells into the tissues owing to a desensitization of the cell surface molecules and the cytokines and their receptors that normally mediate that acute-stress enhancement of cell trafficking and DTH.
Infections and other types of pathogens trigger acute phase responses and activate both innate and adaptive immune mechanisms. Normally, the course of an infection is characterized by a delicate balance between the inflammatory cytokines that activate adaptive immune responses, the innate immune defenses, and the actions of anti-inflammatory factors such as glucocorticoids and anti-inflammatory cytokines that keep the body's own defense from over-reacting and causing damage or death.(19),(20) One type of allostatic state that does result in a measurable allostatic load is the dysregulation of immune system modulators that contributes, along with genetic risk factors, to autoimmune and inflammatory diseases such as multiple sclerosis, rheumatoid arthritis and Type I diabetes.(21) Another related type of allostatic state is the imbalance of immune system regulators that accompanies chronic fatigue syndrome, fibromyalgia and related types of hyperalgesia and chronic pain. (6),(15)
Allostasis and Allostatic Load: Organizing Principles for Neural Control of Body Functions
The paradox of protection and damage also applies to the brain, which we know is the ultimate regulator of body functions. The release and actions of EAAs illustrate the delicate balance between protection and adaptation, on the one hand, and damage on the other hand. Indeed, another example of allostasis is the increase in levels of extracellular glutamate in the hippocampus during restraint stress,(22) which is involved in remodelling of dendrites of hippocampal CA3 pyramidal neurons. We know this because the remodeling is prevented by an NMDA receptor blocker and by phenytoin, a Na+ channel blocker and anti-epileptic drug.(23)
The stress-induced elevation of extracellular glutamate that can be measured by intrahippocampal microdialysis is attenuated by adrenalectomy, suggesting a dependence on adrenal steroids.(22) This mechanism becomes less efficient as rats age and constitutes an example of an allostatic state in which the elevation of a mediator of allostasis fails to go back to baseline when the stressor is finished. When aging rats are subjected to restraint stress and microdialysis, there is an exacerbation of both the level of extracellular glutamate and a prolongation of the response after the stress is terminated.(24) Exacerbation leads to allostatic load at the tissue level, since the elevated glutamate is likely to potentiate the morphological and other effects that occur through activation of NMDA and other excitatory amino acid receptors. Aging rats show increases in Ca++ currents(25),(26) and changes in structure and function related to cytoskeleton and excitatory neurotransmission that may underlie cognitive impairment.(27),(28),(29)
Moreover, excitatory amino acids are also involved in damage caused by seizures and ischemia. Elevated glucocorticoids exacerbate damage caused by ischemia and seizures and amplify the negative effects of excessive EAA. Yet, we have also seen that EAA activity is essential for processes such as learning and memory. The ability to generate and maintain spines on dendrites appears to be essential for compartmentalization of calcium ions during stimulation via NMDA and AMPA receptors and serves a protective role as well as a role for information flow.
Whereas the consequences of the interactions between EAA and glucocorticoids are very clearcut, when it comes to exacerbating damage produced by ischemia and seizures, the effects of the interactions between these mediators in the course of chronic stress are biphasic and generally appear to be protective to some degree, although this is ambiguous as will be discussed further below.
Recap: How the Brain Handles Acute and Chronic Stress
As discussed in a previous Cyberounds® (which you may want to re-read), the nervous system is the interpreter of which events are "stressful" and determines the behavioral and physiological responses to the stressor. The nervous system, similar to the mediators of allostatic load, demonstrates paradoxical biphasic actions. In the brain, strong emotions frequently lead to "flash-bulb" memories -- e.g., where we were and what we were doing when we heard of the September 11, 2001 terrorist attacks at the Pentagon and World Trade Center, or remembering the location and events associated with a very positive life-event, like proposing marriage or receiving a promotion or award.
We saw in the previous Cyberounds® noted above, that both catecholamines, acting via beta adrenergic receptors, and glucocorticoid hormones, acting via intracellular receptors, play an important role in establishing these long-lasting memories. Other brain structures participate along with the autonomic nervous system. The amygdala plays an important role in this type of memory,(30) aided by the autonomic nervous system, which picks up a signal from circulating epinephrine.(31) The process is also facilitated by the hippocampus, which helps us remember "where we were and what we were doing" at the time the amygdala was turned on in such a powerful way.(30),(32)
Thus, epinephrine and glucocorticoids promote the memory of events and situations, which, in the future, may be dangerous, and this is an adaptive and beneficial function. The paradox for the brain comes when there is repeated stress over many days or when glucocorticoid levels remain high because of adrenal overactivity or poor shut off of the stress response or the diurnal rhythm. Then there is atrophy of pyramidal neurons in the hippocampus and dentate gyrus(33) and inhibition of on-going neurogenesis in the dentate gyrus as well as a possible loss of glial cells.(34)
The net result of some or all of the processes is that the hippocampus, as well as the prefrontal cortex and the amgdala, undergoes a shrinkage in size, with impairment of declarative, contextual and spatial memory. As noted in a previous Cyberounds®, this can be picked up in the human brain by neuropsychological testing accompanied by MRI in such conditions as recurrent depressive illness, Cushing's syndrome, post-traumatic stress disorder, mild cognitive impairment in aging and schizophrenia.(35)
Yet the unanswered question is whether these changes represent a prelude to permanent damage or a potentially reversible structural plasticity. On the one hand, after very prolonged periods of chronic psychosocial stress, as in subordinate monkeys living in a dominance hierarchy, pyramidal neurons may actually die.(36) On the other hand, as we have seen in the previous Cyberounds® the hippocampal shrinkage in Cushing's disease is at least partially reversible within several years when the hypercortisolemia is surgically corrected.
What About Permanent Damage, Or Increased Vulnerability From Chronic Stress?
In the rat model of chronic restraint stress (CRS), discussed in the previous Cyberounds®, the stress-induced remodeling of the dendrites in the hippocampus is largely available within 7-10d if CRS is terminated at the end of three weeks,(27) and there are signs that the hippocampus retains a certain degree of resilience after this duration of daily stress (see Figure 1).
Figure 1. Neurons and Protection from Stressors.
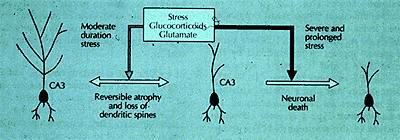
Neurons in the CA3 region of the hippocampus illustrate the paradox of how neurons protect themselves in response to stressors but at the same time may become more vulnerable to damage from stress or from additional trauma. It is very likely that neurons in other brain areas show the same response to stressors, involving glucocorticoids and excitatory amino acids, along with other tissue and hormonal mediators.
Whereas neurogenesis is reduced in the dentate gyrus (DG) and dendrites are shorter and less branched,(23),(38),(39) after three weeks of CRS there is an increase in the expression of polysialated neural cell adhesion molecule (PSA-NCAM) expression consistent with increased mobility of neuronal processes even in the face of reduced DG neuron production. This is because the ability of neuronal processes to expand or contract, and of newly formed neurons to make connections, is dependent on the extracellular environment in which PSA-NCAM plays an important role.(40) PSA-NCAM expression is associated with regions of the brain that show structural plasticity such as the inner granule cell layer of the dentate gyrus and the mossy fiber terminals of CA3.(41)
If three weeks of CRS is accompanied by reversibility of the remodeling of structural plasticity, does prolongation of stress finally lead to damage? The answer is still incomplete and under investigation. However, we know that continuation of CRS for a total of six weeks abolishes the up-regulation of PSA-NCAM noted after three weeks of CRS and results in a significant 6% reduction in DG volume and 13% reduction in granule neuron number.(38) We do not know yet whether structural remodeling occurring after six weeks of CRS are spontaneously reversible, as they appear to be after three weeks of CRS, or whether reversal of remodeling after six weeks of CRS can be accelerated by anti-depressants or anti-epileptic drugs that block the effects of stress and glucocorticoids on remodeling. Nor do we know with certainty whether the structural changes occurring with CRS increase or decrease the vulnerability of the hippocampus to damage by excitotoxicity. We shall now consider arguments and data on both sides of this important question.
Decreased Vulnerability to Damage?
On the side suggesting that CRS might cause some form of limited protection, there is the phenomenon of ischemia preconditioning.(42) What this means is that prior stimulation of the hippocampus by transient ischemia can induce a protective mechanism that may reduce the damage produced by a full-scale ischemic event. It is not clear if the same mechanisms might be operative when stress is applied and whether they might affect the response to excitotoxicity in response to seizures, but this possibility needs to be kept in mind if it turns out that prior CRS has a protective effect on subsequent responses to excitotoxic challenge.
Increased Vulnerability to Damage?
On the other side of the argument, that CRS might increase vulnerability to damage, we have noted earlier in this Cyberounds® series that glucocorticoids exacerbate damage to the hippocampus caused by ischemia(43) and seizures.(44),(45). One possibility is that the immune system is involved. Recent data indicate that glucocorticoids may exacerbate excitotoxic damage, at least in part, by facilitating trafficking of immune cells to the injury cite,(46) where cytotoxic T cells are able to produce cytotoxic death of neurons.(47) A recent study created damage in hippocampal neurons in cell culture and added a mixture of immune cells. When the immune cells can make physical contact after excitotoxic damage is initiated in vitro, they produce excitotoxic effects; these cytotoxic effects do not occur when physical contact between immune cells and neurons is prevented by a membrane, indicating that diffusible substances are not involved.(47)
Yet the brain is considered to be an "immune privileged" organ, and immune reactions normally do not occur. Indeed, there appear to be endogenous protective agents, besides those hypothetically involved in ischemic preconditioning, that are up-regulated in the brain in response to damage or threat of damage. One of the prominent features of excitotoxic damage is the robust induction of calcitonin gene related peptide (CGRP) in terminals and cell bodies in hippocampus and in mossy cells. The increased expression of CGRP in mossy cells is especially prominent after bilateral adrenalectomy (ADX) under conditions in which there is apoptosis (cell death) of granule cells, and the CGRP immunoreactivity is enhanced within the inner third of the molecular layer of the DG.
The neuroimmune peptide, CGRP, is one of the most diverse and influential immunoregulators of the periphery. This important neuropeptide has multiple functions including: its actions as a potent vasodilator(48) and an immune modulator,(49),(50),(51),(52),(53),(54) as well as a neural and immune developmental molecular, a modulator of hormone release involved in growth and development, and a stimulator of sympathetic outflow, which is mediated by CRF and an inducer of apoptosis [reviewed in (55)]. Some of the different functional roles for CGRP may not be independent, but may be part of a cascade of events that constitute the healing response to injury. A number of studies have shown that CGRP is expressed following various kinds of trauma and plays an important role in the acute phase response that may be of particular relevance to the outcome of the regional injury response in the central nervous system.(55),(56).
In recent studies, the expression of CGRP within the hippocampus increases in five separate models of CNS injury: adrenalectomy-induced cell death in dentate gyrus,(57) intrahippocampal colchicine injection,(57) trymethyltin ingestion(58) and kainic acid injections. In each case, the expression of this peptide was limited to the specific region of damage and in association with the surviving neuronal population. Although the up-regulation of CGRP may be associated with neuronal cell survival,(59) other studies have shown that both microglia and astrocytes express CGRP receptors and that exposure to physiological levels of CGRP induces the immediate early gene, c-fos (a sign of rapid activation of genes in cells) in microglia and astrocytes, and increases plasminogen activators.(60) Thus, the role of CGRP may then not only protect against immune system damage to neurons but also participate in plasticity and healing.
Conclusion
In many systems of the body, including the brain, the mediators of adaptation are also the mediators that can cause damage when they are over-produced and dysregulated. Excitatory amino acids working through NMDA, AMPA and metabotropic glutamate receptors illustrate this paradox, and the study of adaptation and damage mediated by EAA receptors constitutes a significant part of basic and applied research in modern neuroscience. See Figure 1.
This Cyberounds® series has summarized how excitatory amino acids work through a variety of ionotropic and metabotropic receptors to mediate the neurotransmission that promotes learning and memory. It has also described the importance and function of dendritic spines in both normal function and as a protective device against damage. NMDA receptors, in particular, are also mediators of the apparently reversible structural plasticity caused by chronic stress, and we have also seen that they play a key role in the damage caused by ischemia and seizures.
Yet, NMDA receptors and EAA do not work alone and the participation of many other endogenous factors and circulating hormones is important to the final outcome. In particular, the acute activation of EAA and NMDA receptors is part of the process (allostasis) that leads to adaptive responses such as synaptic plasticity associated with learning and memory, whereas the excessive activity of EAA and NMDA receptors in the aftermath of a stroke or seizure leads to an allostatic state that can cause wear and tear (allostatic load) and lead to permanent damage.
Glucocorticoids are among the circulating hormones that both facilitate learning during allostasis and yet exacerbate damage after stroke and seizures. Finding ways to control the excesses of both glucocorticoids and NMDA receptor activation without blocking their beneficial effects is a great challenge for the successful development of therapeutic agents.