Course Authors
Howard Eichenbaum, Ph.D.
Dr. Eichenbaum is at the Center for Memory and Brain, Boston University.
Dr. Eichenbaum has been a consultant to Saegis Pharmaceuticals within the past three years.
This activity is made possible by an unrestricted educational grant from Forest Laboratories.
Estimated course time: 1 hour(s).

Albert Einstein College of Medicine – Montefiore Medical Center designates this enduring material activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
In support of improving patient care, this activity has been planned and implemented by Albert Einstein College of Medicine-Montefiore Medical Center and InterMDnet. Albert Einstein College of Medicine – Montefiore Medical Center is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Upon completion of this Cyberounds®, you should be able to:
Describe the physiological requirements for long-term potentiation and long-term depression
Describe the cellular mechanisms by which glutamate mediates long-lasting synaptic plasticity
Describe how AMPA and NMDA receptors contribute to memory.
Glutamate receptors play a pivotal role in the mechanism of synaptic plasticity that underlies memory. This Cyberounds® summarizes our understanding of the role of the excitatory amino acid (EEA) glutamate receptors in a form of synaptic plasticity called long-term potentiation (LTP) and then outlines the evidence that glutamate-mediated LTP is critical to memory.
Hippocampal LTP: Model Memory Mechanism
LTP was discovered in the early 1970s by a Ph.D. student Terje Lomo. He observed that repetitive high frequency electrical stimulation of the pathway from the entorhinal cortex to the dentate gyrus of the hippocampus altered the excitability of this pathway for a prolonged period, leading Lomo to name the phenomenon long-term potentiation. Subsequent research has characterized the basic properties of LTP(11),(13) and generated considerable excitement about this phenomenon as a model for lasting, history-dependent synaptic change.(2),(3),(4),(5),(9)
Five fundamental properties of LTP highlight why it is an attractive cellular model of memory:(49)
- LTP is a prominent feature of the physiology of the hippocampus, a brain structure universally identified with memory.
- LTP develops very rapidly, as one would require of a plausible memory mechanism, typically within one minute after a single stimulus train delivered with the proper parameters.
- Like a strong memory, LTP can be observed for hours after a single stimulation train, or for weeks or more after repetitive stimulations.
- LTP is specific to those synapses activated during the stimulation train; other neighboring synapses, even on the same neurons, are not altered.
- LTP is associative in that potentiation occurs best when multiple inputs are stimulated simultaneously.
The specificity and associativity of LTP have been demonstrated most elegantly in studies that employ activation of separate pathways that synapse on the same hippocampal neurons.(35) In these studies, the two pathways involve the combination of a "weak" input, designated as one that does not produce potentiation at any stimulation level, plus a "strong" input for which a threshold level of stimulation suffices to produce LTP (Figure 1).
Figure 1. Hippocampal LTP.
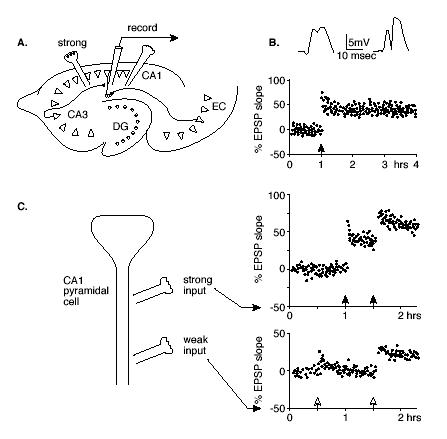
Click to see full sized image
A. Illustration of a horizontal section through the hippocampus showing the pathways by which pyramidal cells in CA1 are stimulated by either a strong input from CA3 or a weak input from the entorhinal cortex (EC).
B. Excitatory postsynaptic potentials (EPSPs) recorded after single pulse stimulations of the strong path before (left) and after (right) high frequency stimulation. Below is the standard method for tracking the changes in the EPSP slope over a period of hours.
C. Associative LTP. A schematic diagram of a CA1 pyramical cell and loci of strong and weak stimulation, and measurements of the EPSP slopes following single pulses of each type of input. The weak input stimulation alone (open arrow at 0.5 hr on lower graph) produces only a transient change, but strong stimulation alone (filled arrow at 1 hr) produces LTP. Combined strong and weak stimulation (both arrows at 1.5 hr) result in LTP at both synaptic sites.
Specificity is observed when high frequency stimulation of the strong pathway produces LTP only at the synapses activated. Associativity is observed when the weak input is activated at the same time as the strong input, resulting in LTP of the weak as well as the strong pathway. Furthermore, even weak LTP can create a synaptic "tag" such that strong activation of a neighboring pathway within a few hours can result in lasting potentiation of both the "strong" pathway and the previously "tagged" synapses.(22),(23) Thus LTP could serve to associate or integrate patterns of activity over a prolonged time window.
Induction of LTP in area CA1 (see Figure 1) requires two fundamental synaptic events -- activation of presynaptic inputs and depolarization of the postsynaptic cell.(10),(12),(15),(37),(50) This combination of events was proposed to be the essential requirements of a synaptic mechanism for associative memory by Hebb(27) and, therefore, this kind of LTP is referred to as "Hebbian." Both events are ordinarily accomplished within a single high frequency stimulus train -- the initial stimulation depolarizes the cell for a relatively prolonged period during which the following stimulations provide simultaneous presynaptic activations. However, high frequency stimulation is not required per se. Instead, for example, direct depolarization of the postsynaptic cell by injection of current through an intracellular electrode, combined with low frequency presynaptic input, will suffice. Conversely, LTP induction can be blocked by preventing depolarization, or by hyperpolarization of the postsynaptic cell.
The molecular mechanism that underlies LTP induction in CA1 involves the special properties of a combination of glutamate receptors (Figure 2).
Figure 2A. Molecular Mechanism of the Induction of LTP.
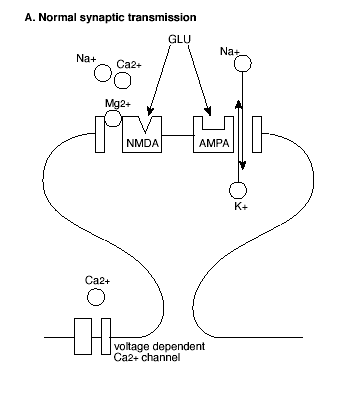
GLU = glutamate; See text for explanation.
Most prominent are the glutamate receptors that are excited by N-methyl-D-aspartate (NMDA receptors) and those that are activated by a-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA receptors). The functions of these receptors in normal synaptic transmission and LTP have been characterized by pharmacological manipulations. NMDA receptors are selectively and competitively blocked by the antagonist D-2-amino-5-phosphonovalerate (AP5). AP5 has little effect on excitatory postsynaptic potentials (EPSPs) elicited by low frequency stimulation, indicating that AMPA receptors, and not NMDA receptors, mediate normal synaptic transmission in the hippocampus. By contrast, AP5 completely blocks LTP following high frequency stimulation trains, indicating that glutamate activation of NMDA receptors is critical to this form of synaptic plasticity.
Figure 2B. Molecular Mechanism of the Induction of LTP.
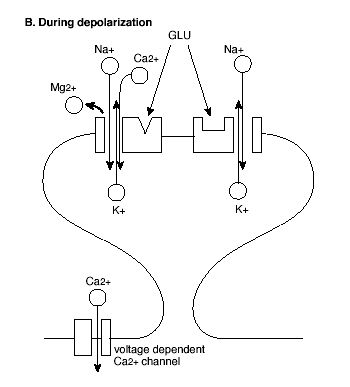
GLU = glutamate; See text for explanation.
However, AMPA and NMDA receptors both play critical roles in the induction of LTP (Figure 2). First, activation of AMPA receptors increases the permeability of the postsynaptic membrane to both sodium (Na+) and potassium (K+) ions, but does not alter cell permeability to calcium (Ca++). By contrast, activation of NMDA receptors increases permeability to Ca++ as well as to Na+ and K+ ions. Second, in contrast to AMPA receptors, ion flow through NMDA receptors is highly dependent on the voltage state of the postsynaptic cell at the time of NMDA receptor activation.
In the resting state, NMDA receptor channels are blocked by another divalent ion, magnesium (Mg++), which prevents the flow of the other ions even in the presence of glutamate at the receptor. However, when the membrane of the postsynaptic cell is depolarized, Mg++ is expelled from the receptor channel, allowing glutamate to bind and the ions including Ca++ to flow. Thus, initially a high frequency stimulus train activates the AMPA receptors, depolarizing the postsynaptic cell membrane. This unblocks the NMDA receptor channels so that succeeding stimuli activate the NMDA receptor, allowing Ca++ to enter the postsynaptic cell.
The entry of Ca++ to the intracellular space is a key step in the induction of LTP. This is shown in at least three lines of work: First, LTP is prevented when Ca++ is bound by intracellular injection of a calcium chelator (that binds calcium); Second, LTP is triggered by intracellular injection of a caged Ca++ compound that releases calcium molecules; Third, the entry of Ca++ into the postsynaptic cell following stimulation trains has been directly observed using a type of microscopy that can track labeled calcium molecules. Glutamate may also activate metabotropic (that is, non-synaptic) receptors coupled to phosphoinositide-mediated release of intracellular stores of Ca++ as an amplification mechanism.
The succeeding steps in the establishment and maintenance of LTP are less well understood. The leading view is that the role of Ca++ is to activate kinases, enzymes that phosphorylate proteins, transforming them into their active configuration. Specific candidates of the critical kinases include type II Ca++/calmodulin-dependent kinase (CaMKII), Ca++/phospholipid-dependent protein kinase C (PKC), and a tyrosine kinase called fyn.
In particular, CaMKII is a very attractive candidate for maintaining synaptic potentiation. It is present in large quantities in the postsynaptic area and, although its initial activation depends on Ca++, it undergoes an autophosphorylation by which it becomes independent of the transient Ca++ influx. This prolonged activation mediates long-lasting consequences, one of which might be the conversion of inactive AMPA receptors into active ones, "waking up" previously "silent" synapses.(31),(32),(38),(40),(41)
LTP may also be maintained by structural changes, including increased synaptic contact size and density.(26),(42) The functional significance of this structural change was demonstrated in a study that examined a form of learning that depends on the hippocampus. On each training trial, rats were presented with a brief noise, followed by a blank "trace" interval and then a mild electrical shock delivered around the eye, causing a blink. After several training trials, rats learned to blink during the trace interval in anticipation of the delayed shock. In animals that learned, the density of CA1 synaptic contacts was increased, and this training-induced structural change was prevented by blocking NMDA receptors.(34)
If there was only a form of plasticity that increased synaptic efficacy, eventually all synapses would become "saturated", i.e., raised to a ceiling level of efficacy, and no further learning could occur. So, in addition to the potentiation of synapses, there must be a mechanism of de-potentiation or long-term depression (LTD) of synaptic efficacy. Moreover, LTD can enhance the relative effect of LTP at particular synapses by decreasing the excitability of neighboring synapses, thus improving signal-to-noise of the information code at the potentiated synapses. Also, combinations of LTP and LTD at different synapses of the same cell increases the range of possible information coding patterns for the cell over the number of patterns that can be generated by LTP alone.
Several variants of LTD have been observed.(7),(8),(14),(39) The form of lasting hippocampal synaptic depression that has received the most attention is homosynaptic LTD. Homosynaptic LTD involves depression of synapses where presynaptic activity occurs in the absence of postsynaptic firing. This is usually accomplished by stimulating the inputs to a cell at a very low frequency. At low stimulation frequency, some excitatory synapses are activated, but the cell is very unlikely to fire at that time because the stimulation also activates surrounding inhibitory elements that hyperpolarize the cell. This results in the combination of presynaptic activation and postsynaptic hyperpolarization, the opposite of the Hebbian requirement. In the typical protocol, the same pathways used for study of LTP in CA1 are depressed by a long series of continuous 1-Hz stimulation, and this form of synaptic plasticity shares several features with LTP. Homosynaptic LTD is reversible by LTP-inducing stimulation. It is also input-specific and requires activation of NMDA receptors as well as a rise in Ca++ concentration in the postsynaptic cell.
Lisman(36) proposed a model for the signaling cascades that might mediate LTP versus LTD, which depends on the timing of activations and consequent rising levels of intracellular Ca++. According to this model, large rises in Ca++ level that would accompany rapid trains of stimulation activate CaMKII or other protein kinases, whereas smaller rises in Ca++ would activate a protein phosphatase, such as calcineurin. The kinase and phosphatase mediators could act on a common pathway that alters synaptic proteins by regulating the phosphorylation of synaptic proteins (i.e. enhanced).
Beyond the Hippocampus: LTP and LTD
LTP has been demonstrated in widespread areas of the brain and is viewed by many as a universal plasticity mechanism. Among the areas where LTP, and/or LTD, have been demonstrated include the neocortex, piriform cortex, amygdala, neostriatum, cerebellum and spinal cord.
Perhaps best studied of the non-hippocampal areas is the visual cortex.(6),(7),(57) Stimulation of the input layer IV of the visual cortex results in LTP and LTD in principal cells of Layer III with the same protocols effective for producing these phenomena in the hippocampus.(1) It is interesting to note that while high frequency stimulation produced LTP, low frequency stimulation resulted in LTD. Both forms of synaptic modification are synapse-specific and depend on NMDA receptors. Intracellular injections of electrical current that produce postsynaptic depolarization or hyperpolarization paired with low frequency synaptic activation produce synaptic enhancements or decrements, respectively.(21)
Based on studies of both the hippocampus and visual cortex, Bear has proposed that the balance of LTP and LTD may be regulated by a common learning rule. According to his model, there is a "modification threshold" such that active synapses are potentiated when the total synaptic response exceeds a particular threshold and depressed when the total synaptic response falls below that threshold. Furthermore, the threshold for directions of modification can change or "slide" with overall activity levels of the system.
A major example of the "sliding" modification threshold is the change in modifiability of the visual cortex during development. Early in postnatal development, when the eyes are first exposed to patterned illumination, cells in the visual cortex are quite sensitive to competing inputs from the two eyes, and rapidly develop "ocular dominance" patterns of differential responsiveness to the two eyes; these patterns support stereoscopic vision.
After an exposure period of several months, the threshold for establishment of ocular dominance is considerably reduced. This phenomenon has clinical significance in cases of strabismus or "lazy eye", wherein the cells of visual cortex do not receive the normal competitive inputs from the two eyes. If the problem is fixed early, the still low modification threshold allows proper generation of the capacity for stereoscopic vision. However, if the treatment is delayed beyond the critical period, the modification threshold for the visual cells becomes too high, precluding establishment of the normal ocular dominance patterns that support stereoscopic vision.
Glutamate-mediated LTP and Memory
As Stevens(61) put it, LTP is so attractive that it would be a shame if the mechanism underlying LTP turned out not to be a memory mechanism. Many studies have sought to connect the mechanisms of LTP and memory, and in particular the mechanisms involving glutamate receptors.(43),(44),(52) Here I will outline a few of those studies.
As discussed above, LTP is mediated by activation of AMPA receptors, and unmasking of AMPA receptors may maintain LTP. Correspondingly, AMPA receptors are indeed critical for memory. However, the specific role of AMPA receptors in memory per se is not clear-cut because manipulation of AMPA receptors affects neural communication in addition to neural plasticity. Nevertheless, some studies have shown specific memory associated affects of AMPA receptor manipulations. For example, mice lacking AMPA receptor subunit GluR-1 are impaired in memory for recent visits to locations in a maze, but can learn a consistent location over many trials, indicating a selective role for AMPA receptors in memory for unique episodes.(53) Conversely, AMPA receptor facilitation enhances memory in several situations.(25),(54),(58)
The most compelling data on a potential connection between the molecular basis of NMDA-dependent LTP and memory have come from experiments where NMDA anatagonists or genetic manipulation of NMDA receptors are used to influence LTP and, correspondingly, affect learning. The earliest and strongest evidence supporting a connection between LTP and memory came from studies on spatial learning by Morris and colleagues.(47)
Morris developed a maze learning task requiring hippocampal function. In this task, the maze involves a swimming pool in which the water is made opaque by the addition of a milky powder, and an escape platform is submerged just under the water at a predetermined location. Rats are good swimmers but prefer to find and climb onto the platform. Typically, they are trained to find the platform from any of four starting positions around the periphery of the maze, and they show learning by shortening the time required to escape from all starting points. At the end of training, their memory is assessed using a probe test -- the platform is removed and rats exhibit good memory by swimming in the vicinity of the former escape locus.
Initially, Morris and his colleagues showed that AP5-induced blockade of NMDA receptors prevents new spatial learning in the water maze. Drug-treated rats swim normally but do not reach the same level of rapid escape as normal rats. Indeed, the drug-treated rats often adopt a strategy of swimming at a particular distance from the walls of the maze. This reduces the time required to find the escape platform to some extent without knowing the exact location of the platform. Thus NMDA receptors are required specifically for memory of the platform location independent of the development of an adaptive swimming strategy.
In the probe tests, normal rats show a distinct preference for swimming in the vicinity of the former escape locus but drug-treated rats show little or no such bias, indicating the absence of memory for the escape location. By contrast, AP5 has no effect on remembering the escape locus when training was accomplished prior to drug treatment. The intact ability to recall a normally acquired memory is fully expected because NMDA receptors are required only for the induction of LTP and not for its maintenance.
Additional research has also shown how NMDA receptor-dependent LTP might play a continuing role in updating one's memories.(60) To accomplish this, the researchers developed a version of the water maze task in which the location of the escape platform is moved every day and animals are given four (4) trials to learn the new location (Figure 3).
Figure 3. One-trial Place Learning in the Morris Water Maze Task.
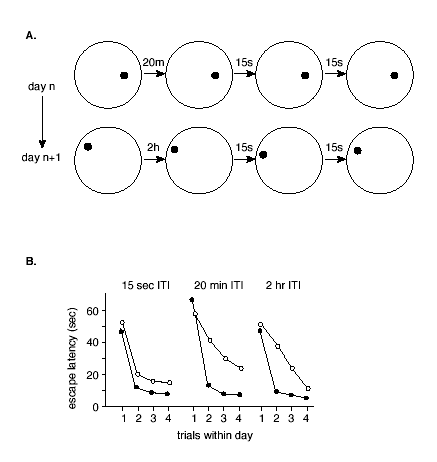
A. Example test sessions. On each session the animal is given four (4) trials with the escape platform in a novel location. Note that the first inter-trial interval is variable, and the others are constant at 15 sec.
B. Effects of infusion of AP5 directly into the hippocampus. After initial training on the task, rats treated with AP5 or placebo were tested on a series of 4-trial sessions with the escape platform in a novel location each day. On some test sessions, the interval between trials 1 and 2 was 15 sec. On these sessions, rats given AP5 performed as well as normal subjects in showing a substantial reduction in the latency that indicated intact memory. On other sessions, the interval between trials 1 and 2 was 20 min or 2 hr. On these sessions, normal animals also showed good retention, but AP5 rats showed substantially less reduction in their escape latencies, indicating memory impairment. The later inter-trial intervals were all 15 sec, and all animals show substantial latency decreases over these brief intervals. Filled circles = controls; open circles = AP5.(data from 60).
Rats became skilled at the task such that they consistently found the platform very rapidly -- on the second trial. Subsequently, animals were tested with different memory delays inserted between the first and second trial on each day. Infusion of AP5 into the hippocampus prior to learning AP5 treatment resulted in a deficit on trial 2 performance. Moreover, this deficit was dependent on the time interval between trial 1 and trial 2, such that no impairment was observed with a 15 second inter-trial interval but significant deficits ensued if the inter-trial interval was extended to 20 minutes or longer.
These data suggest that memory for specific episodes of spatial learning remains dependent on NMDA receptors and LTP even after the animals have learned the environment and the general rules of the spatial task. The generality of this finding has been extended to a critical role for NMDA receptors in memory for a particular location where a specific food was once found.(20)
Other research has used targeted genetic manipulations to show that blocking NMDA-dependent LTP also results in severe memory impairments.(63) A particularly impressive example involved a knockout limited to post-development activation of the genes for the NMDA receptor specifically in the CA1 subfield of the hippocampus that selectively blocks LTP in that region.(64),(65) Mice with this highly selective temporal and anatomical restriction in NR1 function are severely deficient in water maze learning as well as in other types of memory dependent on hippocampal function.(28),(51),(55) A complementary study examined the effects of a mutation that results in over-expression of the NMDA NR2B receptor, producing an enhanced activation of NR1 receptors. This mutation facilitates LTP and, correspondingly, improves several kinds of memory dependent on the hippocampus.(62)
Emerging evidence suggests that NMDA receptors in specific subregions of the hippocampus may mediate different aspects of memory. For example, mice with a knockout of the NMDA receptor in CA3, unlike those with the knockout in CA1, can learn the standard water maze task. However, they are impaired when a subset of the spatial cues are removed(48) and fail in the version of the task where the mice must learn a new escape location each test day.(49) Another study using pharmacological blockade distinguished a critical role for CA3 NMDA receptors in learning new spatial environments as contrasted with a critical role for CA1 NMDA receptors in maintaining spatial memories for an extended duration.(33)
Plasticity of Hippocampal Neuronal Firing Patterns
Research on the nature and persistence of spatial representations of single hippocampal neurons and neuronal populations in animals with compromised capacity for LTP further complements the evidence involving NMDA receptors in memory. These studies involve extracellular recordings from so-called "place cells," hippocampal principal neurons in CA1 and CA3 that fire when the animal is in a particular location in its environment. These firing patterns are thought to reflect memories of experiences in the spatial environment.
Acute pharmacological blockade of NMDA receptors does not prevent the initial establishment of new spatial representations.(29) Nor does NMDA blockade prevent the expression of previously established spatial representations. However, new spatial representations established during the NMDA blockade are not persistent; they last briefly, then new representations are formed when the rat is re-introduced into the seemingly familiar environment. The rapid loss of spatial representations corresponds to the rapid deterioration of spatial memory observed when NMDA receptors are blocked.(60)
Another study examined the consequences of knockout of CA1 NR1 receptors for spatial firing patterns in groups of neighboring hippocampal CA1 neurons in mice. Hippocampal cells in these mice had diminished spatial specificity and a reduction in the coordinated activity of neurons tuned to overlapping spatial locations.(45) Furthermore, the findings on place cells were associated with the spatial memory impairment such that the loss of coordinated activity in mutant hippocampal place cells leads to a poorer prediction of sequential locations during navigational behavior. These results suggest that the network processes that bind together neuronal representations of spatial cues are particularly dependent on LTP.
LTP and Memory Outside the Hippocampus
Additional studies suggest that NMDA-dependent LTP may also mediate plasticity in other brain areas that support memory. A particularly good example of this work involves a set of studies that examined taste learning mediated by the gustatory cortex of rats.(56) When rats are exposed to a novel taste and subsequently become ill, they develop a conditioned aversion specifically to that taste, and this learning is known to depend on the gustatory cortex. Blockade of NMDA receptors by infusion of the antagonist AP5 produces an impairment in taste aversion learning, whereas the same injections given prior to testing a previously established memory, or into an adjacent cortical area, had no effect. Thus NMDA-dependent LTP underlies modifications of cortical taste representations that support taste memory.
Also, an extensive line of evidence demonstrates a critical role for NMDA receptors in the amygdala, a brain region associated with fear and anxiety, that support a form of emotional memory called fear potentiated startle.(16),(17),(18),(19) In this task, animals are initially exposed to pairings of a light or a tone with foot shock. Subsequently, their reflexive startle response to a loud noise is considerably augmented or "potentiated" in the presence of the conditioned light or tone. Infusion of the NMDA receptor blocker AP5 into the amygdala prior to training prevents conditioning, but similar treatment after conditioning or prior to testing has no effect on later expression of potentiated acoustic startle. Thus, NMDA-dependent plasticity in the amygdala is critical for the development of fear conditioning but not for the expression of already learned fear responses.
Further evidence distinguished the role of NMDA receptors in fear memory as opposed to a possible direct involvement of NMDA receptors in the expression of conditioned fear.(24) Rats were first conditioned to fear one stimulus (e.g., a light) by presenting the stimulus paired with shock. Subsequently, they were trained to fear a second stimulus (e.g., a tone) by pairing the stimulus (the tone) with the conditioning stimulus they had already learned to fear (the light). AP5 infused into the amygdala during the second conditioning stage actually enhanced the fear expression to the initial conditioning stimulus, while at the same time preventing the second stage of conditioning. This finding strongly supports the conclusion that NMDA receptors are differentially involved in the plasticity of startle but not in the performance of a potentiated startle response.
Similar results have also been obtained in another fear conditioning task that involved inhibition of approach to a location previously associated with shock.(30) AP5 had no effect on initial learning but blocked the ability of the animal to maintain the memory. By contrast to the effects of AP5, injection of the AMPA receptor antagonist CNQX into the amygdala blocks the expression of fear potentiated startle. This pattern of data is entirely consistent with our understanding of the differential roles of NMDA and AMPA receptors respectively in the establishment of synaptic plasticity and in its expression, highlighting the role of glutamate receptors in synaptic plasticity in the amygdala as critical to fear memory.
Conclusions
In mammalian systems, the most popular model for the cellular and molecular mechanisms that underlie memory is long-term potentiation (LTP) and its sister phenomenon long-term depression (LTD). Both phenomena follow Hebb's rule -- increases in synaptic efficacy (facilitation of synaptic transmission) marking LTP occur as a consequence of repeated activation of a presynaptic element and its participation in the success in firing the postsynaptic cell, whereas decreases in synaptic efficacy (decrements in synaptic transmission) marking LTD occur as a consequence of the absence of correspondence between activation of a presynaptic element and postsynaptic cell activation.
An understanding of the molecular and cellular mechanisms of some forms of LTP is emerging. The induction of one prominent form of LTP involves the combined activation of multiple glutamate receptors, which occurs when AMPA receptors are initially activated to depolarize the post-synaptic cell, causing the release of a magnesium block of the NMDA receptor and thus allowing the transmitter glutamate to activate that receptor. The now unblocked magnesium channel is a route for an influx of calcium, which begins a molecular cascade of events that both stabilizes the changes in the post-synaptic cell and induces gene expression and permanent cellular modifications that maintain enhanced connectivity.
There are compelling lines of evidence favoring the view that LTP and memory share common cellular mechanisms and, in particular, share a dependence on glutamate receptors. Across a broad range of learning tasks and brain systems, blocking LTP with drugs or genetic manipulations that affect glutamate receptors results in a pattern of amnesia reflected both in memory impairments and instability of the relevant neural representations. Conversely, enhancement of glutamate dependent LTP facilitates memory.