Course Authors
Bruce S. McEwen, Ph.D.
Release Date: 05/08/2002
Upon completion of this Cyberounds®, you should be able to:
Discuss the structure and function of the hippocampal region of the brain
Describe how aging of the brain, especially the hippocampus, is assessed
Discuss the evidence for and against neuronal loss in the aging brain.
Introduction
We are acutely aware of the aging process in ourselves and in our friends and loved ones and cognizant of the considerable individual differences in the rates of aging. This is particularly true for those aspects of aging which impair mental and physical functioning. How much of this is in our genes and how much is environmentally driven? There is no simple answer but it is important to understand how our experiences can influence the rate at which we age, i.e., the "weathering" of our bodies and our brains.
A recent article in the New England Journal of Medicine provided data that a lifetime of experience of economic hardship resulted in earlier declines in physical functioning, cognitive performance and mental health.(1) The concept of allostatic load,(2) which was developed in four previous Cyberounds® conferences, provides the basis for understanding how stress hormones can produce wear and tear on the body and brain.
It is the brain that is particularly important for the aging process because it controls neuroendocrine and neural processes that affect metabolism, cardiovascular and immune function. Yet, the nervous system lacks the ability to generate new nerve cells. Once enough neurons die there is a permanent loss of function. However, there is controversy concerning how many neurons die in the aging brain. There are, moreover, alternative explanations for loss of function in the aging brain that are not so drastic and are potentially treatable.
This Cyberounds® is the first of four that addresses some of the current information and controversies regarding the "weathering" of the brain. The series focuses on a region of the brain, the hippocampus, which is particularly sensitive to the aging process, in part because it is very sensitive to the effects of stress. This Cyberounds® considers how we assess aging and whether the changes seen in animals and humans in cognitive function are necessarily due to permanent loss of nerve cells.
Importance of the Hippocampus
The hippocampus is a particularly vulnerable and sensitive region of the brain that is also very important for declarative and spatial learning and memory.(3) Input to the hippocampus comes from the entorhinal cortex and this input is the first to degenerate in the early stages of Alzheimer's disease.(4) The cross-sectional 'tri-synaptic' organization of the hippocampus is shown in Figure 1.
Figure 1. Tri-Synaptic Organization of the Hippocampus.
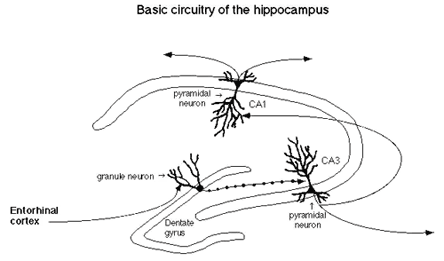
The first synapses are those from the entorhinal cortex input to the dentate gyrus; the second consist of the dentate gyrus projection to the CA3 neurons; while the third synapses are those from the CA3 to the CA1 neurons. Through this tri-synaptic circuit flows information from the cortex which is processed and distributed to other brain regions, including the subcortical regions of the brain.(5)
Hippocampal neurons are like high-performance race cars -- they have enormous capabilities in terms of their activity and plasticity but, as a result of these characteristics, are particularly vulnerable to seizures, strokes and head trauma, as well as stressful experiences.(6),(8) Their remarkable plasticity is demonstrated by long-term synaptic potentiation and depression, dendritic remodelling, synaptic turnover and neurogenesis in the case of the dentate gyrus.(7),(9)
The work of aus der Muhlen and Ockenfels(10) first drew attention to potentially toxic actions of adrenal steroids. These investigators reported darkly stained neurons in the hippocampus of guinea pigs exposed to high levels of glucocorticoids, an observation that has been confirmed and extended to repeated stress in subsequent studies,(11),(12) although there are still some doubts as to whether 'dark neurons' may be artifacts of tissue trauma.(13) In 1968, we discovered receptors for adrenal steroids in the hippocampus(14) and it is now known that two types of adrenal steroid receptors exist in the hippocampus and other brain regions and mediate a variety of adrenal steroid effects on excitability, neurochemistry and structure.(8)
Phillip Landfield(15) and then Robert Sapolsky(16),(17) provided evidence for a role of adrenal steroids in neuronal aging in the hippocampus, leading to the formulation of the "glucocorticoid cascade hypothesis."(6) This hypothesis states that glucocorticoids participate in a feed-forward cascade of effects on the brain and body in which progressive glucocorticoid-induced damage to the hippocampus promotes progressive elevation of adrenal steroids and dysregulation of the hypothalamo-pituitary-adrenal (HPA) axis.(6) According to this view, "weathering" is a process that can accelerate with increasing dysregulation of the HPA axis and provides a gradually increasing allostatic load.
Subsequent work has provided considerable support for the model for both brain and body aging and has extended our knowledge of the aging hippocampus from animal models to humans.(18),(19),(20) At the same time, the new information has revealed a number of complications and problems with the original formulation.
First, finding neuronal death in the hippocampus during aging is technically very problematic. Recent information from stereological cell counting has down-played the importance of neuronal death, as opposed to declining neuronal function and various forms of structural plasticity. This will be discussed in this Cyberounds®.
Second, the role of the hippocampus in HPA regulation is more complex than originally believed, considering both its neuroanatomical connections to the hypothalamus and the nature of negative feedback regulation of the HPA axis. Moreover, there is a life-long pattern of reactivity of the HPA axis which helps to set the rate of 'weathering" of the brain and body. This will be discussed in Part II of this Cyberounds® series.
Third, the hippocampus is a dynamic and plastic region of the adult as well as developing brain, in which stress hormones (glucocorticoids) have an important role; yet, glucocorticoids do not act alone on the hippocampus and there is new information down-playing the relative importance of adrenal steroids, as opposed to excitatory amino acids and other modulators, including neurotrophins and calcium ions, as well as sex hormones. This will be the subject of the third Cyberounds® in this series.
Fourth, our new appreciation of the plasticity of the hippocampus has opened the way to possible treatment strategies to reverse hippocampal atrophy and retard the onset and progression of Alzheimer's disease. This will be discussed in the fourth Cyberounds®.
Obtaining an Accurate Description of the Aging Brain and Body
Chronological age alone is not sufficient to predict the state of the aging brain and body. The problem that individual differences poses for aging research is that individual animals or human subjects must be evaluated for cognitive and physical status, relative to other individuals of the same age, in order to understand the meaning of any single physiological, neuroanatomical, neurochemical or molecular measure.(21) For the hippocampus, as well as for the rest of the body, additional information is needed regarding both cognitive function and stress mediators (i.e., HPA and sympathetic nervous system reactivity), as well as measures of body function that are influenced by these mediators. This is true not only for animal models(22) but also for studies on humans, for which longitudinal information about HPA activity and other measures of allostatic load(2) in aging subjects have been shown to predict cognitive decline and onset of cardiovascular disease.(23),(24),(25) Thus, the most meaningful studies have been done comparing cognitively-impaired and -unimpaired groups of aging individuals and in groups of individuals differing in their levels of allostatic load.(23),(24),(25) We shall return to the topic of individual differences in Part II.
Neuron Death Is Not Inevitable or the Only Cause of Age-related Impairment
There has been a tendency to interpret the "glucocorticoid cascade hypothesis" as implying that hippocampal neuron death is an inevitable consequence of brain aging and that age-related impairments in cognitive function are solely related to such neuronal loss. This notion has given away to a more flexible view of brain aging, in which impairments in hippocampal functioning can be studied in terms of potentially reversible, as well as irreversible, changes in neuronal structure, neurochemistry and function.(26),(27)
The first challenge to the notion of neuronal loss is from a methodological problem in estimating neuron number in the brain. Because of the gradual time course of brain aging, even in rapidly-aging small animals like rats, observing neuronal death by counting dying neurons is futile because it would have to be such a slow process to account for the gradual changes in function. For that reason, studies of pyramidal neuron damage and the role and mechanism of action of glucocorticoids and excitatory amino acids have utilized kainic acid damage or transient ischemia models.
Even while the issue of age-related neuronal loss is controversial, and by no means resolved, there is evidence that the aging hippocampus undergoes progressive changes with age in calcium homeostasis, the plasticity of response to glucocorticoids and in the expression of markers related to neuroprotection and damage. The activity of L-type calcium channels undergoes an increase in hippocampal CA1 pyramidal neurons of aging rats and results in an increased after-hyperpolarization(28) which alters the electrophysiological properties of these neurons and makes them more vulnerable to damage. In cultured embryonic hippocampal neurons that are maintained for 28d, there is an increase in calcium channel activity and in after-hyperpolarization that is accompanied by decreased neuronal survival; blocking L-type calcium channels increased neuronal survival.(33) It is interesting to note that the increased after-hyperpolarization is associated with an enhanced induction of long-term depression (LTD) in CA1 pyramidal neurons and an impaired induction of long-term potentiation (LTP).(34) Thus, insofar as LTP and LTD may be related to synaptic plasticity during learning,(35) these age-related changes suggest a possible basis for cognitive impairment in aging rats.(34)
Glucocorticoids enhance calcium channel activity and after-hyperpolarization(36) and glucocorticoid receptor expression shows a progressive failure of negative feedback regulation in old versus young rats. In young rats, repeated stress causes a down-regulation of glucocorticoid receptor levels, thus decreasing glucocorticoid efficacy on various target genes, whereas, with increasing age, this down-regulation is lost, thus preserving glucocorticoid actions.(29) This natural mechanism in the young hippocampus (repeated stress ---> reduced magnitude of the glucocorticoid feedback signal ---> reduced impact of glucocorticoids on calcium channel activity, among other effects) may be protective, for increased calcium channel activity contributes to free radical generation and other processes that may damage neurons.(37),(38) With the loss of stress-induced down-regulation of glucocorticoid receptors, older rats appear to lose this protective device and may be more vulnerable to increased levels of glucocorticoids which do appear to accompany aging, particularly in cognitively-impaired rats.(29)
Even if outright neuronal loss is not a major event in the aging hippocampus of cognitively-impaired rats, there are indications that gene products associated with neurodegeneration and damage are differentially regulated in the aging-impaired brain, compared to unimpaired aging rats and young rats, although the interpretation of the results is very complex.(39) In aging, cognitively-impaired rats, the levels of mRNA for the 695
amino acid form of the beta amyloid precursor protein (betaAPP) and for the magnesium-dependent superoxide dismutase (Mg-SOD) were both elevated throughout the hippocampus compared with young rats at the same time the levels of the betaAPP protein and Mg-SOD protein were both depressed.
Another aspect of the aging hippocampus is alteration in glutamate release associated with an age-related increase in dynorphin content of the hippocampus.(41) Dynorphin is a peptide that was originally identified as one of the "endogenous opioids" and which now is known to function as a neurotransmitter in some nerve cells. Dynorphin appears to be co-present in mossy fiber nerve terminals with glutamate, and is release by these terminals during nerve stimulation. The age-related increase in dynorphin content changes is accompanied by impairments of spatial learning, something that is increasingly evident in some rats more than others as they get older. Since dynorphin is present in the mossy fiber pathway as a co-transmitter with glutamate, the increased levels of this peptide may have an inhibitory autoregulatory function at the mossy fiber synapse,(41) resulting in less glutamate release and impairing the synaptic transmission and plasticity that is essential for learning and memory.
Conclusions
What a few years ago was thought to be an open-and-shut case of age- related and stress-induced neuronal loss has turned out to be far more complicated and interesting as far as the multiple processes that appear to be involved in the aging of the hippocampus. Indeed, neuronal loss with aging may have been over-emphasized, so much so that the irreversibility of the age-related changes is called into question. There are a number of aspects of these new findings that will be addressed in the next three Cyberounds®. The first issue is the role of the HPA axis and glucocorticoids as the basis for the "weathering" of the hippocampus and as the reason for individual differences in the rate of "weathering," a subject we will discuss in Part II of this series.
Acknowledgments
Research in the author's laboratory on some of the topics discussed in this article is supported by NIH Grants NS07080 and MH41256 and by the Health Foundation (New York), Servier (France) and UCB (Belgium).