Course Authors
Robert G. Lerner, M.D.
Release Date: 06/30/1997
Upon completion of this Cyberounds®, you should be able to:
State the physiological mechanism which produces sickling
Describe the pain management guidelines for sickle cell crises
Describe the newer alternative therapies for reducing vasco-occlusive crises.
A 25-year-old black man first came to the attention of our hematology group at the age of 19. At that time he presented to another hospital with priapism, which failed to improve after treatment with hydration and analgesics. He informed us that he had sickle cell anemia which was diagnosed at birth because both his parents had sickle trait and they wanted him tested. (Parents are often confused about screening for sickle cell disease, and the NIH has provided a useful guide designed for use by parents that can be accessed at http://isis.nlm.nih.gov/ahcpr/sickle/www/scdptxt.html.)
A companion guideline for use by physicians is also available here.
Additional family history is that a sister died at the age of 23. She apparently went to bed after taking analgesics for abdominal pain that was thought to be a sickle cell crisis. She was found dead in bed and an autopsy revealed a ruptured ectopic pregnancy. This patient's childhood was not marked by severe disease, although he had multiple episodes of aches and pains. His first hospital admissions for sickle cell crisis occurred at age 18. When he came to our hospital his priapism had persisted for three days and was still very painful. He was treated with exchange transfusion replacing his sickle cells containing S hemoglobin with normal cells containing A hemoglobin. Within 24 hours he had partial detumescence, but to this day the distal half of the shaft of his penis remains enlarged except for the glans which is normal in size.
He remained asymptomatic for several months but then developed multiple sickle cell crises marked by abdominal and back pain requiring hospital admission and treatment with analgesics. At the age of 21 he developed left hip pain. X-ray films revealed bilateral aseptic necrosis of the femoral heads, worse on the left, and not present on films done at the age of 19. The pain decreased and he now has occasional pain, not requiring analgesics, and walks without an obvious limp. At the age of 23 he had a recurrence of priapism which regressed promptly after treatment with exchange transfusion. At the age of 24, he developed an ulceration on the skin over the medial malleolus of the left ankle which healed over several months with conservative treatment, only to recur three months later.
Figure 1. Typical Leg Ulcers in Sickle Cell Anemia.

Figure 1 and Figure 2 from the American Society of Hematology Slide Bank. Used with permission.
In October of 1996, oral hydroxyurea was started and the ulcer healed. He has had no episodes of crisis pain since and has remained only mildly symptomatic.
Sickle cell anemia is the most clinically significant of a group of diseases in which sickle hemoglobin is present. It was first described by James Herrick.(1)
Figure 2. Three Views of the Peripheral Blood Smear of Sickle Cell Anemia with Red Blood Cell Showing Typical Sickle Shape.
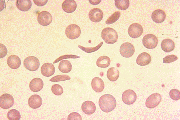


Linus Pauling deduced the genetic nature of the disease and the relationship between hemoglobin deoxygenation and sickling.(2) An online database cataloging human genetic diseases including hemoglobinopathies is available at http://www3.ncbi.nlm.nih.gov/Omim/.
This patient's experience brings up several issues about the treatment of sickle cell disease. Although he has not had serious problems with the management of sickle pain, it is a problem which deserves special mention. Sickle cell crises usually occur unpredictably and those that result in hospitalization average five to seven days duration. There is no specific therapy and treatment is usually analgesia, hydration and treatment of complications, if present.
In every hospital where I have worked, the pattern of narcotic analgesic use has been to start with doses of meperidine that are inadequate and of insufficient frequency. Many sickle cell anemia patients have life-long experience with this approach and state their expectations when arriving at emergency departments. They may ask to be started on 100 to 200 mg of meperidine, only to find that the emergency department staff believe that they are addicts and start with lower doses.
Meperidine is not recommended as a first line narcotic for sickle crisis. Meperidine has a short half-life and often must be given every two hours. At the same time, it is metabolized to normeperidine, which has a longer half-life, and accumulates to toxic levels causing agitation and even seizures. When narcotics are needed, morphine or hydromorphone are better choices. The Agency for Health Care Policy and Research has developed pain management guidelines that are very useful. They should be individualized according to the patient's past experience.
Exchange transfusion therapy, which we used for priapism in this case, is particularly effective for serious acute events such as acute chest syndrome. The acute chest syndrome is a leading cause of death and although previously thought to be clearly different from pneumonia, may be precipitated by infection as well as thromboembolic or fat-embolic disease. Exchange transfusion, using pheresis machines which are now widely available, is often the most expeditious way to carry out the transfusion. Matching for the most common minor blood groups, as well ABO matching, should be done to avoid alloimmunization. Chronic transfusion therapy can reduce the frequency of painful vaso-occlusive crises, stroke, acute chest syndrome and bacterial infection(3) but carries the risks of iron overload, blood borne illness and the other problems of transfusion dependence.
Newer alternatives to chronic transfusion can also reduce the incidence of painful vaso-occlusive crises. Bone marrow transplantation can be curative but carries the risk of early death due to the procedure, as well as graft versus host disease, late malignancy, sterility, neurologic and other complications.(4)
Oral hydroxyurea is much more easily used. This agent is one of several that are known to increase fetal hemoglobin levels in sickle cell anemia patients, and this was the basis for the clinical trials that have established that it is clinically effective in decreasing the incidence of crises.(6) There are some doubts about whether this is the entire explanation.(7) First, the data did not show that the decrease in incidence of crises correlated with the increase in fetal hemoglobin. The decrease in crises occurred even before the fetal hemoglobin rose. The best correlation appeared to be with the decrease in white blood cell count. Additionally, some patients were intolerant to even small doses of hydroxyurea, while others tolerated large doses without improvement. Current recommendations are to start at doses of 15-20 mg/kg/day and monitor closely for myelotoxicity (absolute neutrophil count <2,000/mm3 or platelet count <80,000/mm3 or hemoglobin falling more than 2 g/dl) so that drug can be stopped. Hydroxyurea is believed to be leukemogenic when used in patients with polycythemia vera, and the incidence of late malignancy is unknown, as is the risk to pregnant patients. Despite these caveats, the results of the trial were so striking that an alert was sent over the Internet to Medline users by the NIH when the data were first released. The full text of the alert can be accessed at http://www.nlm.nih.gov/databases/alerts/sickle_cell.html. Many unknowns are ahead of us but it is very gratifying to finally be able to offer some effective therapy for sickle cell anemia.