Course Authors
Arindam Singha, M.D., and Stephen E. Kirkby, M.D.
Release Date: 09/10/2018
Upon completion of this Cyberounds®, you should be able to:
Discuss the basic pathogenesis of cystic fibrosis, and recent trends in survival and outcome;
List the diagnostic criteria including CFTR mutation;
Describe the organ specific manifestation of disease;
Manage both acute exacerbation and chronic disease including pharmacologic and non pharmacologic therapies;
Evaluate and apply the new and emerging disease-modifying therapies for CF.
Cystic fibrosis (CF) is an autosomal recessive disease caused by genetic mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) protein. At the cellular level, the CFTR protein is an anion channel normally present in the epithelial membrane of many cells. A dysfunctional or deficiency in the CFTR protein results in abnormal ion gradients, especially in chloride, and bicarbonate ions.
The downstream effects of the abnormal ion gradients include dehydration on the airway surface due to an excess of sodium and water reabsorption, and abnormally low pH of the liquid in the airways, which denatures the endogenous antimicrobial agents and causes unusually thick mucus that is unsuitable for normal mucociliary transport.(26)
In a healthy lung, inhaled bacteria are killed by antimicrobial properties of the airway surface liquid, and then cleared by the mucociliary transport. In patients with CF, however, this process is less effective leading to a vulnerable host defense system where bacteria can proliferate. As a result, chronic and recurrent bacterial airway infections, chronic inflammation with mucus plugging of airways and progressive bronchiectasis represent the hallmark of pulmonary CF. While chronic lung disease is the primary cause of morbidity and mortality, over the past decades, it has become evident that CF is an illness that affects the whole body and mind, not just the lungs.
Epidemiology
CF is most common in the populations of northern European ancestry. Birth prevalence varies among people of different ethnic backgrounds, and also from country to country. The disease occurs in 1 in 3000 in white Americans, 1 in 4000-10,000 Latin American and 1 in 15,000–20,000 African Americans. CF is relatively uncommon in Africa and Asia.(19)
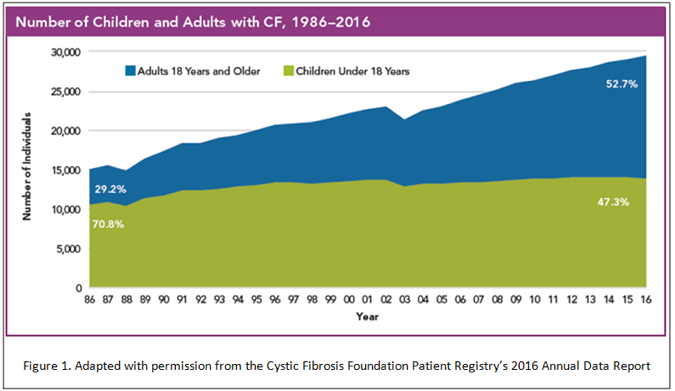
Although CF is a disease typically diagnosed in childhood, it is rapidly becoming an adult predominant disease. In 2016, adults made up 52.7% of the CF population, compared to 29.2% in 1986 (Figure 1). It is important for an internist, therefore, to be familiar with the diagnosis and chronic management of CF patients. With the advent and widespread use of newborn screening, late diagnosis of CF will likely become less common, which only highlights the importance of keeping a keen index of suspicion for the atypical presentation of CF.

Figure 1. Number of Children and Adults with CF, 1986-2016.
Diagnosis
Early diagnosis of CF is key, as early intervention leads to better lung function and nutritional status in CF.(32) Today, most new diagnoses of CF occur at the newborn screening. In 2016, 62.4% of total diagnoses and 86% of diagnoses in patient younger than six months of age were detected through the newborn screening.(2)
Diagnosis of CF consists of finding specific clinical (phenotypic) characteristics in combination with biochemical or genetic markers of CFTR protein dysfunction.
Both of the following criteria must be met for diagnosis:(10)
1. Clinical symptoms consistent with CF in at least one organ system (explained below)
AND
2. Evidence of CFTR protein dysfunction based on:
- Elevated sweat chloride level (>60 mmol/L) on two occasions;
- Presence of two disease-causing mutations in CFTR, one from each parent;
- Abnormal nasal potential difference.
Measurement of nasal potential difference is a test predominantly used in research. It involves inserting a nasal catheter and measuring the electrical response of the nasal mucosa to perfusion of different ionic solutions. The test is labor intensive, technically difficult and is not readily available in all CF centers and, therefore, is typically not the first line diagnostic method of choice in most clinical practice. It is important to note that these tests should be run in a CF center where the laboratory technicians are well versed in the assays, and the tests are done routinely and frequently.
Diagnostic criteria are the same for adults and children, although the presentation in adults may vary from patient to patient, and diagnosis later in life typically is associated with a better prognosis. A physician should have high index of suspicion for CF when adults present with chronic or recurrent pancreatitis, chronic sinusitis, unexplained bronchiectasis, chronic cough with sputum production, especially in the presence of chronic pseudomonas infection, allergic bronchopulmonary aspergillosis (particularly in the absence of asthma) and male urogenital issues such as azoospermia. Diagnosis of CF follows a strict guideline, and if the criteria are not met it is important to look for alternate causes such as primary ciliary dyskinesia or immunodeficiencies.
Manifestation of Disease
Pulmonary
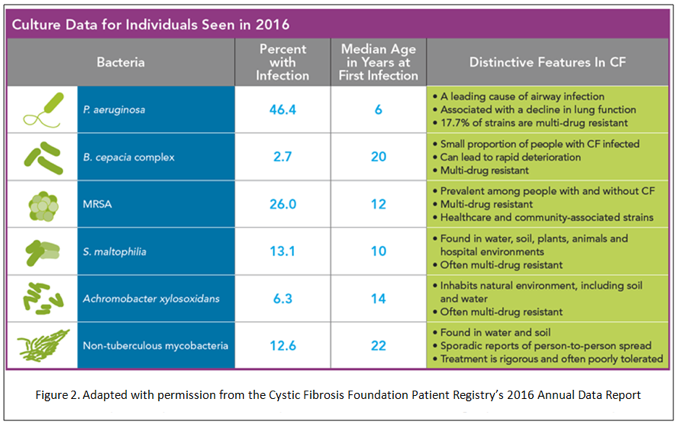
Inflammation, recurrent infections, mucus plugging and development of bronchiectasis are the hallmark of pulmonary CF. Pulmonary complications remain the leading cause of death and accounted for two-thirds of the deaths among CF patients in 2016.(2)(5)In CF patients, the respiratory tract is quickly colonized with various pathogenic organisms soon after birth and infection. Figure 2 outlines the various organisms cultured from lower respiratory tract of CF patients.
Figure 2. Culture Data for Individuals Seen in 2016.
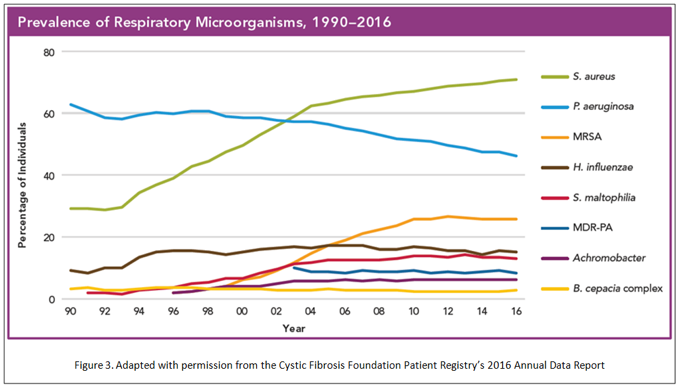
Infections with Pseudomonas aeruginosa is a characteristic feature of CF and is a cause of significant morbidity. Although developments of antipseudomonal antibiotics, including inhaled agents, have decreased the incidence of pseudomonas infection (Figure 3), the incidence of multi drug resistance pseudomonas remains unchanged. Burkholderia cepacia complex, Staphylococcus species (MSSA and MRSA), Stenotropomonas multiophila and non-tuberculous mycobacteria are also common virulent pathogen in CF.
Figure 3. Prevalence of Respiratory Microorganisms, 1990-2016.
Burkholderia cepacia is especially associated with rapid decline in lung function and increased mortality. Allergic bronchopulmonary aspergillosis (ABPA), an intense allergic reaction to Aspergillus fumigatus, is a well-known complication in CF patients. It presents with cough, central bronchiectasis, infiltrates on chest imaging, peripheral eosinophilia and elevated serum IgE level.
Gastrontestinal
It is hypothesized that heterozygous mutation of the CFTR protein provided a survival advantage during the cholera epidemic in 19th century Europe.(7)(11) Just as it regulates the chloride and fluid secretion in the bronchial epithelial cell, the CFTR protein can be found in the epithelium of the intestines, pancreatic duct and the biliary system.(11)(31) While heterozygosity may prevent against secretory diarrhea, homozygous mutation of the CFTR gene leads to thickened secretions which occlude hollow structures in the GI system. In the pancreas, for example, thickened secretions leads to obstruction, recurrent acute inflammation and ultimately pancreatic insufficiency.(11)(12)(14) In the same manner, thick luminal mucus of the GI tract and decreased motility can produce intestinal obstruction.(19) Obstruction of intrahepatic bile ducts can cause focal biliary cirrhosis as well.
Abdominal pain is a common complaint in CF patients. In addition to the typical causes of abdominal pain in the average population, several other etiologies such as constipation and distal intestinal obstruction (DIOS), pancreatitis and GERD can be more common in CF patients.(31) Most patient with CF (>90%) will have pancreatic insufficiency and will require pancreatic enzyme supplementation. Symptoms of pancreatic insufficiency include diarrhea, foul-smelling greasy stool, weight loss or poor weight gain, flatus, abdominal discomfort and fat soluble vitamin deficiency. Chronic recurrent pancreatitis in a healthy adult, especially in the absence of heavy alcohol consumption or gall stones, should prompt an evaluation for CF.
Endocrine, Nutrition and Bone
CF-related diabetes mellitus (CFRD) is a unique disorder with clinical features of both type 1 and type 2 diabetes. The pathogenesis is complex but is believed to be due to fibrosis and replacement of pancreatic tissue with fat from chronic inflammation, leading to insufficient insulin secretion. Furthermore, a component of insulin resistance from chronic infections is also thought to play a role. The incidence of CFRD progressively increases from adolescent to adulthood and many patients with mild disease may go undiagnosed. Prevalence as high as 43% in adults over 30 years of age has been reported in one study.(17)(18)(32)
There are several features unique to diabetes in a CF patient: increased energy expenditure, chronic recurrent infections, abnormal liver function, abnormal intestinal transit and absorption cause unpredictable levels of serum glucose and insulin. Most CF patients tend to have some preserved basal insulin production, thus making fasting hypoglycemia less severe and ketoacidosis a relatively rare phenomenon. Postprandial hyperglycemia appears to be a prominent feature however.(32)
Hemoglobin A1c (HbA1c) is not a reliable marker of disease control in CFRD, as patients can still have unacceptably high postprandial hyperglycemia despite a "good" HbA1c. The CF Foundation recommends, therefore, that all patients be screened for CFRD by means of an annual oral glucose tolerance test. CFRD is associated with more severe lung disease, increased pulmonary exacerbations and overall poor nutrition, and is an independent risk factor for poor outcome in CF patients.(13)(32) Any CF patients with unexplained weight loss or a sudden drop in pulmonary function should be evaluated for CFRD.
Pathogenesis of low bone mineral density in CF is a complex phenomenon that involves both accelerated bone turnover and slow bone growth. Poor GI absorption of calcium and vitamin D, diminished sunlight exposures and hypogonadism if present slow bone growth in CF patients. Simultaneously, repeated corticosteroid exposure, diminished physical activity and chronic pulmonary inflammation result in accelerated bone breakdown. Exposures to immunosuppressant agents post transplant further hastens bone breakdown.(32) The prevalence of osteoporosis in adult CF patients varies from 38-77%.(25) Adults should be screened with DEXA, and if bone mineral density is normal repeat screening should take place every two to five years.
Adequate nutrition and preventing weight loss are associated with increased long-term survival and good lung health. Patients should be encouraged to consume a high-calorie, high-fat diet. Patients should have regular visits with dieticians well versed in managing CF-related nutrition to learn about energy dense foods. Most CF patients need supplementation with fat soluble vitamins (A, D, E, K). Despite the addition of enzyme supplement, dietary education and oral supplementation of vitamins, gastrostomy (PEG) tube feeding may be necessary to maintain healthy BMI. The CF Foundation recommends targeting a BMI >23 for men and >22 for women.
Mental Health
Like most chronic illnesses, there is a high rate of both depression and anxiety in CF patients and their caregivers. The prevalence of depression ranges from 13-33% in adults; and the prevalence of anxiety ranges from 30-33%.(20) Mental illness in CF has been associated with decreased pulmonary function, lower BMI, worse adherence to treatment, more frequent hospitalizations and increased healthcare costs.(20) All CF patients should undergo periodic screening for depression and anxiety.
Multidisciplinary Management of CF
Management of CF patients is best done with a multidisciplinary approach. A CF care team should involve physicians, nurses, respiratory therapists, dieticians and social workers, ideally all trained in caring for CF patients.
Acute Pulmonary Exacerbations
What constitutes an acute exacerbation is not very well defined. Most practitioners would agree that increased cough, changes in sputum color or quality, weight loss or changes in appetite, changes in respiratory exam including presence of new wheezing, or crackles or new radiographic evidence of consolidation are alarming features and should prompt further evaluation.
Aggressive airway clearance and antibiotics based on sputum bacterial cultures are the mainstays of therapy during acute exacerbations. Since the vast majority of adults with CF are infected with pseudomonas, two intravenous antipseudomonal antibiotics are typically given for at least 14 days.(19) Antibiotics are usually combined with aggressive airway clearance techniques (chest physiotherapy, hypertonic nebulized saline,(4) nebulized bronchodilators), three to four times daily, with the goal to not only improve symptoms but also to return to the individual patient's baseline FEV1 value.
Chronic Pulmonary Management
Table 1. The Foundation of Chronic CF Therapy.
| Daily airway clearance |
| Mucolytic therapy with dornase alfa |
| Nebulized hypertonic saline |
| Chronic suppressive antibiotic therapy |
| High calorie nutrition, replacement of pancreatic enzymes and fat soluble vitamins |
| Exercise |
Most significant advancements have taken place in the chronic management of CF. Although the advent of modern antibiotics has made a tremendous impact on overall survival; antibiotics are but a single feature of a successful CF treatment regimen. Dornase alfa, a recombinant human deoxyribonuclease (DNase), cleaves the extracellular DNA found within the airway, thus reducing mucus viscosity and improving mucocilliary clearance when combined with airway clearance techniques such as flutter device, vest therapy or manual percussion.(3)(6) Nearly all airway clearance techniques rely on oscillation either orally in case of the flutter device or by chest wall vibration in case of manual percussion and the vest therapy.
The flutter valve is a hand-held device that the patient blows through to create vibration within the airways. This vibration, accompanied by resistance within the device, helps to mobilize secretions and assists clearance. In the vest therapy, an air pulse generator rapidly inflates and deflates the vest thereby gently compressing and releasing the chest wall several times per second. The vibration at the chest wall is transmitted to the airways which mimics the physiology of coughing.(6) This helps mobilize mucus to the larger airways. Studies have demonstrated that addition of nebulized hypertonic (7%) saline twice daily in patients with CF not only improves FEV1 but also leads to fewer pulmonary exacerbations.(3)(4)
Chronic suppressive antibiotic therapy has become a mainstay in CF therapy and aerosolized antibiotics are of particular interest. The advantage of inhaled antibiotics is high airway concentrations in the lung with minimal systemic side effects. Inhaled tobramycin is the most thoroughly studied of the agents available. In patients with moderate to severe disease and chronic pseudomonas colonization, when given in 28-day on-off cycles, inhaled tobramycin leads to improvement in pulmonary function, decreased pseudomonas burden in sputum culture and reduced hospitalizations.(15)(22)(32) In pseudomonas positive patients, addition of three times a week azithromycin to the standard regimen has also shown improvement in FEV1 and reduction in pulmonary exacerbations when compared to placebo.(15)(23)
Management of CF-related Diabetes
Diabetes in CF patients is managed differently from the standard of care offered patients with type 1 or type 2 diabetic. Oral agents are usually not recommended and short-acting insulin with meals is typically the mainstay of therapy. (16)Some patients do need long-acting basal insulin in addition to the short-acting insulin therapy. Patients with CF tend to have difficulty maintaining weight and so carbohydrate restriction is typically not recommended. Instead patients are encouraged to consume a predictable amount of carbohydrate or meticulously count their carbohydrate intake to match short-acting insulin dosing appropriately. All patients with CFRD should be screened annually for microvascular complications with a dilated eye evaluation and a urinalysis for microalbumin measurement.(16) Diabetic nephropathy is of high concern in CF patients, as most of these patients are exposed to nephrotoxic antimicrobial agents during the course of their lives.
Management of Osteoporosis
Treatment of existing bone mineral disease in CF patients is not well studied. There is some evidence showing that pamidronate may be beneficial.(1) Patients should be encouraged to perform regular weight-bearing exercise, get exposure to sunlight to promote vitamin D formation, maintain a good nutritional status to maintain or increase bone mineral density. Evaluation by an endocrinologist to rule out sex hormone deficiencies or other pre-disposing conditions is also prudent.
Management of Mental Health Issues
CF patients should receive annual screening for depression and anxiety. Behavior therapy including cognitive behavioral therapy (CBT) for depression and exposure-based CBT for anxiety are first-line therapy. Selective serotonin reuptake inhibitors (SSRIs) including citalopram, escitalopram, sertraline and fluoxetine are appropriate first-line antidepressants.(20)(32)
Novel Disease Modifying Therapies
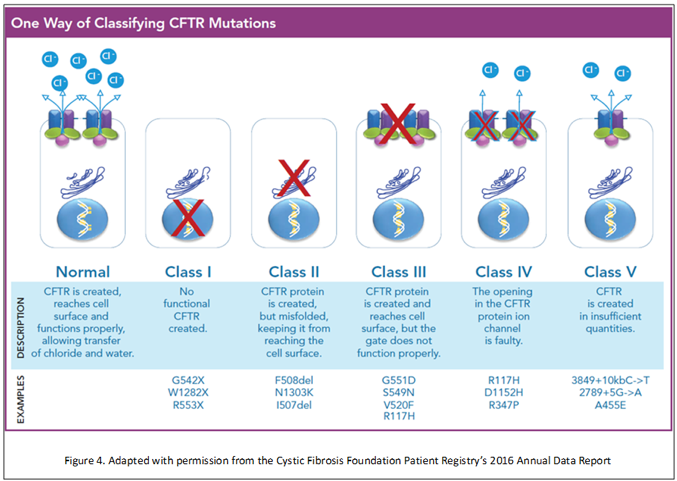
Most CF therapies until now have focused on tackling the secondary downstream effects of the defective CFTR gene. Recently, significant advancements in disease modifying therapies targeting specific mutations in CFTR protein have shown some very promising results. CFTR mutations can be classified in one of five classes (Figure 4). In the most common type of CFTR mutations, Class II, mRNA is translated into a misfolded form of CFTR protein that never reaches the cell surface. A class III mutation leads to CFTR proteins that reach the cell surface, but the chloride channel appears to be dysfunctional.
Figure 4. One Way of Classifying CFTR Mutations.
Ivacaftor is an orally bioavailable agent that targets Class III mutations. It is designed to be a "potentiator," to increase the time that activated CFTR channels at the cell surface remain open, thus promoting chloride transport. When added to standard of care, patients receiving ivacaftor had improved pulmonary function, reduced pulmonary exacerbation (by 55%), increased weight, increased self reported quality of life markers and improved sweat chloride concentration compared to placebo.(15)(21)
Lumacaftor was designed as a "corrector" agent to target Class II mutations. It repairs CFTR misprocessing and increases the amount of functional CFTR proteins at the cell surface. A phase 2 clinical trial as single agent in adults did not show much clinical benefit aside from reduced sweat chloride level. However, when combined with ivacaftor, lumacaftor/ivacaftor leads to increased FEV1, reduced rate of pulmonary exacerbations, reduced rate of hospitalizations, decreased need for IV antibiotics and improvement in BMI.(29)
Despite the promising results seen with lumacaftor/ivacaftor, it is not for everyone. Several observational studies have documented intolerance to lumacaftor/ivacaftor due to adverse respiratory side effects.(8)(9) In addition, strong cytochrome P-450-3A induction by lumacaftor causes prohibitive drug–drug interactions in some patients.(27)
The latest addition to the arsenal in the treatment of CF is tezacaftor/ivacaftor. Like its predecessor lumacaftor/ivacaftor, it is also a combination agent and is approved for patients 12 years of age or older who were homozygous for the Phe508del CFTR mutation. A multicenter, multinational phase 3 trial involving over 500 patients showed promising results.(27) When compared to placebo, patients treated with tezacaftor/ivacaftor had significant improvement in FEV1, reduced rate of pulmonary exacerbations and required fewer hospitalizations or intravenous antibiotics. It was also well tolerated, with rates of adverse effects and discontinuation of drug due to adverse effect similar to placebo.(27)
Lung Transplantation
Lung transplant is an established therapy for end stage cystic fibrosis. CF is the most common reason for lung transplant in patients under 50 and patients, accounting for about 16% of all lung transplant recipients between 1995 and 2015; about 97% of them received bilateral transplants.(24)(33) CF patients have the highest median survival among lung transplant recipients.(34) According to the International Society for Heart and Lung Transplant (ISHLT) registry, the overall 5-year survival among CF patients is 62%.(33) The Canadian CF Registry described a slightly higher 5-year survival rate of 67.7 %.(28)
The ISHLT recommends referral for transplant based on several criteria including pulmonary function, clinical decline and the presence of pulmonary hypertension (Table 2).
Table 2. Timing of Referral for Transplant as Outlined by ISHLT.(30)
| FEV1 | <30% or if rapidly declining despite optimal medical therapy |
| Six-minute walk test | <400 meters |
| Pulmonary hypertension | Mean pulmonary artery pressure >25 mm Hg on right heart catheterization or right ventricular systolic pressure >35 on echocardiogram measured not during acute exacerbation |
| Clinical decline |
??c Pneumothorax ??c Life threatening hemoptysis refractory to bronchial artery embolization ??c Acute respiratory failure requiring non invasive ventilation ??c Worsening nutritional status despite adequate supplementation ??c Increasing antibiotic resistance |
The ISHLT also outlined several clinical and social factors, which pose absolute and relative contraindications to transplant, including recent malignancy, infection with particularly virulent organisms, medical non-compliance and lack of social support (Table 3).
Table 3. Contraindications to Lung Transplant as Outlined by ISHLT.(30)
| Absolute Contraindications | Relative Contraindications |
|---|---|
| Recent malignancy | Age >65 |
| Untreatable significant dysfunction of another major organ system (e.g., heart, liver, kidney, or brain) unless combined organ transplantation can be performed | Severe malnutrition |
| Atherosclerotic disease not amenable to revascularization | Atherosclerotic disease burden sufficient to put the candidate at risk for end-organ disease after lung transplantation |
| Acute medical instability (acute sepsis, myocardial infarction, liver failure, etc.) | HIV infection (Lung transplant can be considered in patients with controlled disease with undetectable HIV-RNA, and compliant on combined anti-retroviral therapy. The most suitable candidates should have no current AIDS–defining illness.) |
| Uncorrectable bleeding disorder | Severe, symptomatic osteoporosis |
| Chronic infection with highly virulent and/or resistant organisms that are poorly controlled pre-transplant | Colonization or infection with highly resistant or highly virulent bacteria, fungi, and certain strains of mycobacteria |
| Active Mycobacterium tuberculosis infection | Hepatitis B/C infection (Lung transplant can be considered in patients without significant clinical, radiologic, or biochemical signs of cirrhosis or portal hypertension and who are stable on appropriate therapy.) |
| Significant chest wall or spinal deformity expected to cause severe restriction after transplantation | Extensive prior chest surgery with lung resection |
| Class II or III obesity (body mass index [BMI] ??JPY35.0 kg/m2) | Class I obesity (BMI 30.0–34.9 kg/m2) |
| Current or repeated/prolonged history of non-adherence to medical therapy | Infection with Burkholderia cenocepacia, Burkholderia gladioli, and multi-drug–resistant Mycobacterium abscessus if the infection is sufficiently treated preoperatively and there is a reasonable expectation for adequate control postoperatively |
| Psychiatric or psychological conditions associated with inability to comply with medical therapy or cooperate with medical professionals | Medical conditions that have not resulted in end organ damage but are not optimally treated (e.g., diabetes mellitus, hypertension, epilepsy, peptic ulcer disease or GERD) |
| Lack of social support | |
| Severely limited functional status with poor rehabilitation potential | |
| Substance abuse/dependence |
Conclusion
CF is becoming a disease of adult medicine. Adults with CF can live normal lives, have normal jobs and start a family. According to the CF Patient Registry's 2016 report, about two-thirds of adults with CF are either studying or working.(2) The number of CF adults who are married or with college degrees has nearly doubled in the last decade as well. Remarkable progress has been made in the acute and chronic management of CF. With the advent of the target-specific therapy we no longer just have to settle for managing the downstream effects of CFTR mutations. Nevertheless, it is important to appreciate that while available therapies have made significant impact on both symptoms and pulmonary function, the disease still poses tremendous treatment burden to the patients and their families.