Course Authors
Siamak Moghadam-Kia, M.D., Rohit Aggarwal, M.D. and Chester V. Oddis, M.D.
Release Date: 12/04/2017
Upon completion of this Cyberounds®, you should be able to:
Discuss the role of systemic glucocorticoid therapy which is considered the mainstay of initial treatment of idiopathic inflammatory myopathies;
Describe the role of methotrexate or azathioprine, other immunosuppressive, immunomodulatory agents or biologic drugs that can be sequentially used alone or in various combinations in the management of polymyositis and dermatomyositis;
List the mechanism of action and common side effects of immunosuppressive or immunomodulatory therapies used in myositis treatment.
Other than glucocorticoids and Acthar, the FDA has not approved any medications for the treatment of myositis.
The idiopathic inflammatory myopathies (IIMs) are a group of heterogeneous, systemic rheumatic diseases that include adult polymyositis (PM), adult dermatomyositis (DM), myositis associated with other systemic autoimmune rheumatic diseases or cancer, juvenile myositis (juvenile dermatomyositis and juvenile polymyositis), inclusion body myositis (IBM) and necrotizing autoimmune myopathy.
Because of their low incidence and prevalence, their variable clinical phenotypes and the small number of randomized, double-blind, clinical trials,(1)(2)(3)(4) the treatment of IIM has been very challenging. Traditional treatment approaches include glucocorticoids and conventional immunosuppressive or immunomodulatory agents such as methotrexate, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus and IVIg.
There has been growing interest in assessing novel and targeted therapies such as biologics that target various pathways implicated in the etiopathogenesis of myositis. Biomarkers involved in the pathogenesis of IIM have been explored using cytokine/chemokine analyses, microarrays and RNA-sequencing analysis, advanced immunohistochemistry and flow cytometry.
Novel classification schemes for IIM based on serologic and histopathologic features may also enhance the design of clinical trials and subject enrollment. (5)(6) Additionally, in the past several years, consensus and data-driven core set measures (CSM) have replaced poorly-standardized muscle strength and functional assessments for evaluation of myositis disease activity and damage. In particular, two international groups, the International Myositis Assessment and Clinical Studies (IMACS) Group and the Pediatric Rheumatology International Trials Organization (PRINTO), have defined and validated consensus CSM for adult and pediatric populations.(7)(8)(9) These consensus outcome measures, along with active international initiatives to develop both data- and consensus-driven response criteria, will assist in studying novel therapies in a more systematic and rigorous fashion.(10)
In this Cyberounds®, we review the conventional and novel immunomodulatory and immunosuppressive treatment approaches used for treating PM and DM, necrotizing autoimmune myopathy or overlap syndromes, and myositis-associated interstitial lung disease (ILD).

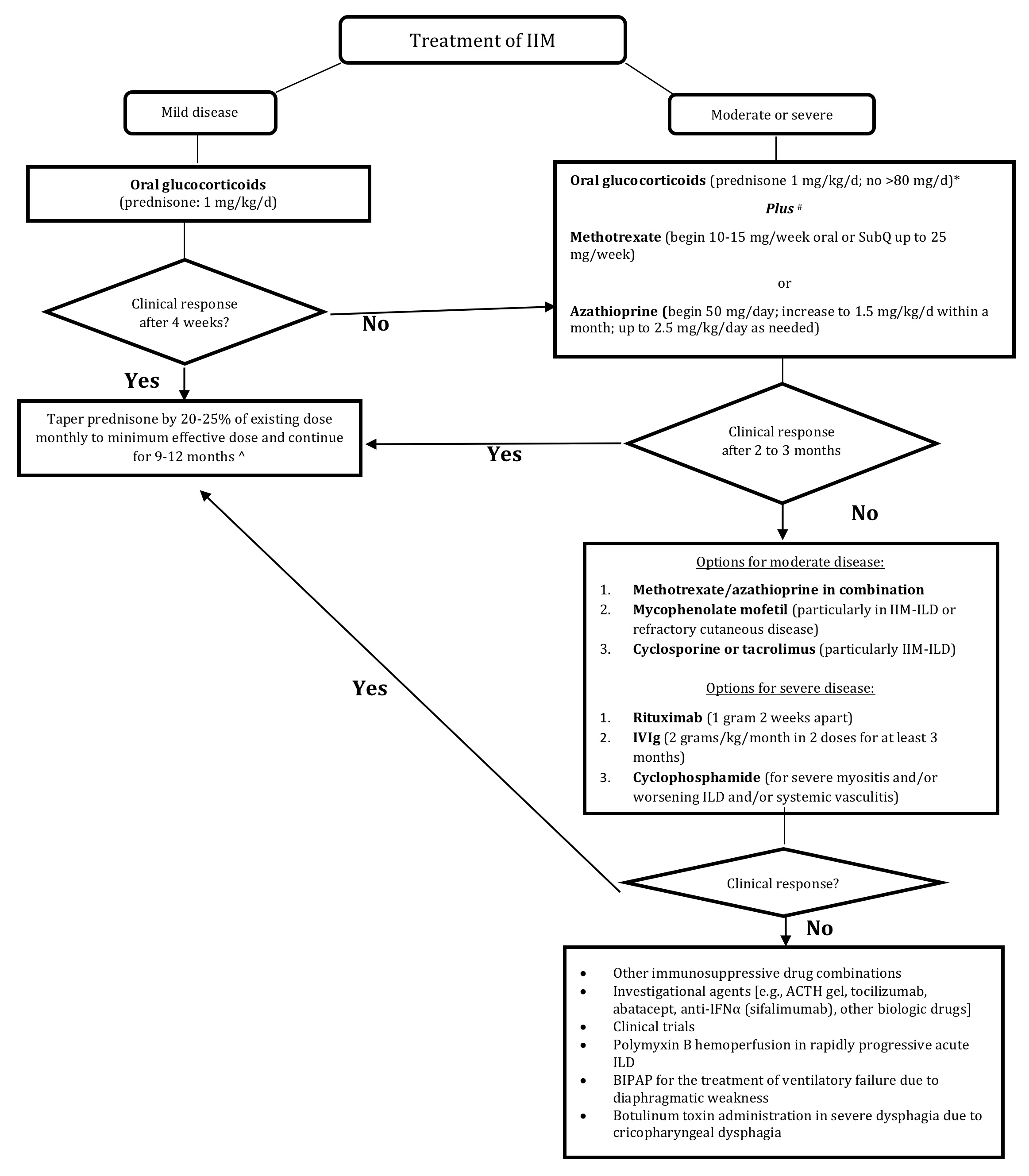
Figure 1. Therapeutic Approach to Idiopathic Inflammatory Myopathy [IIM].
Glucocorticoid Therapy
Despite the lack of placebo-controlled clinical trials, glucocorticoids remain the mainstay of initial treatment of IIM. Glucocorticoids can normalize serum muscle enzymes and improve or preserve muscle strength(11) and are generally initiated with prednisone at a dose of 1 mg/kg/day in divided doses (generally not exceeding 80 mg daily). After four to six weeks, the prednisone dose is slowly tapered to the minimum effective dose using the general guideline of a 20-25% drop every month until the daily prednisone dose reaches 5-10 mg/day. The tapering is then frequently held for total duration of glucocorticoid therapy of at least 9 to 12 months.
Patients with severe disease manifestations, such as marked weakness, severe dysphagia or rapidly progressive ILD, generally receive pulse intravenous methylprednisolone (usually 1000 mg daily for three consecutive days) in combination with another immunosuppressive (IS) agent.(12)
At least half the patients do not respond to glucocorticoid monotherapy.(5) In a retrospective cohort study of 113 patients among those who responded to glucocorticoid alone, most did not regain normal muscle strength and/or flared when steroids were tapered.(11) Some patients with mild myositis can be treated with glucocorticoids alone but most patients require another IS or immunomodulatory drug due to refractory disease, repeated disease flares or to reduce the dose and duration of concomitant glucocorticoid therapy along with associated side effects. Regardless of the choice of initial treatment, early therapy lessens subsequent muscle damage leading to less weakness.(11)
A lack of initial response or worsening while on glucocorticoid therapy requires a re-assessment of the accuracy of the myositis diagnosis. Glucocorticoid-related myopathy or an unrecognized malignancy must also be considered. Repeat muscle biopsy may be necessary to confirm the diagnosis or evaluate glucocorticoid-related myopathy. In the follow-up assessment of IIM patients, improved muscle strength is a more clinically reliable indicator of treatment response than the decline or normalization of the serum muscle enzymes such as creatine kinase (CK). However, in many instances the CK remains an excellent biomarker of IIM disease activity.
Methotrexate and Azathioprine
The first-line conventional immunosuppressive agent for IIM is generally methotrexate or azathioprine. Methotrexate is a folate antimetabolite that irreversibly bind to and inhibits dihydrofolate reductase, resulting in inhibition of DNA synthesis, repair and replication. There are no placebo-controlled clinical trials assessing methotrexate in PM or DM. However, a randomized, open-label, assessor-blind, international, multi-center clinical trial was recently completed in Europe to assess the efficacy and safety of combined methotrexate/glucocorticoid therapy as compared with glucocorticoid therapy alone (ClinicalTrials.gov identifier NCT00651040).(13)
In another recent multicenter, randomized trial in new-onset juvenile DM, 139 patients were randomly allocated to prednisone alone or in combination with either methotrexate or cyclosporine.(14) Combined treatment with prednisone and either cyclosporine or methotrexate was more effective than prednisone alone — at month 6, 24 (51%) of 47 patients on prednisone, 32 (70%) of 46 patients on prednisone plus cyclosporine, and 33 (72%) of 46 patients on prednisone plus methotrexate achieved a juvenile dermatomyositis PRINTO 20 improvement (p=0.0228).
Several retrospective studies have suggested the efficacy of methotrexate in myositis patients, even those refractory to initial glucocorticoid monotherapy.(11)(15) A cohort study of 55 glucocorticoid-refractory patients, reported that methotrexate use was associated with a partial response in 31 and a complete response in nine.(11) Methotrexate can be administered orally or subcutaneously and the dose is often titrated up to 25 mg/week. Methotrexate toxicity monitoring includes hematologic monitoring with assessment of liver enzymes and renal function. Methotrexate pulmonary toxicity leading to pneumonitis is rare but may present a diagnostic challenge in myositis patients with underlying ILD, so methotrexate needs to be used cautiously in the setting of active ILD.(16)
Azathioprine is a derivative of mercaptopurine that inhibits purine metabolism, interfering with cellular replication. In a randomized, controlled, double-blind trial, 16 patients with PM received 60 mg of prednisone daily plus either azathioprine (2 mg/kg/day) or placebo for a period of three months.(2) There was no difference in muscle strength or CK between the two groups after three months, but patients treated with combination therapy had better functional status and required less prednisone for maintenance three years later.(17)
Some retrospective case series have suggested efficacy for azathioprine in treating IIM-ILD(18)(19) including a series of 70 patients with IIM-ILD where azathioprine use in 25 patients led to improvement. Controlled comparisons have suggested that azathioprine and methotrexate have similar efficacy.(11)(20) In a more recent study, survival was higher between five and 10 years in the methotrexate-treated group as compared with those receiving azathioprine, but this could not be confirmed in multivariable modeling for the full follow-up period.(21) In contrast, a retrospective Asian study in PM and DM patients, non-use of azathioprine was associated with higher mortality.(22) Azathioprine is preferred in patients with liver disease, those unwilling to abstain from alcohol or those with IIM-ILD.
Azathioprine is given orally starting at 50 mg per day with dose escalation by 25-50 mg increments every one to two weeks up to 1.5 mg/kg/day. If the response is inadequate after two to three months of therapy, the dose can be increased up to 2.0 or 2.5 mg/kg/day. The azathioprine target dose is lower in patients with renal insufficiency. Monitoring parameters should assess bone marrow suppression, liver enzyme abnormalities and renal dysfunction. Azathioprine toxicity also includes flu-like reactions with fever, gastrointestinal symptoms and pancreatitis. The U.S. Food and Drug Administration (FDA) recommends screening for thiopurine methyltransferase (TPMT) deficiency prior to the initiation of treatment with azathioprine.
In a crossover study, thirty patients with refractory IIM, including those who previously had inadequate treatment responses to either methotrexate or azathioprine alone, were randomized to begin either a combination of weekly oral methotrexate and daily azathioprine or intravenous methotrexate with leucovorin rescue every two weeks for six months. Intention-to-treat analysis showed a trend in favor of those patients who first received the combination therapy.(23)
Mycophenolate Mofetil
Mycophenolate mofetil (MMF), a prodrug of mycophenolic acid, is a reversible inhibitor of inosine monophosphate dehydrogenase, which inhibits guanosine nucleotide synthesis, resulting in inhibition of T and B lymphocyte proliferation. MMF use in IIM has been reported in several small case series.(24)(25)(26)(27) In an open study in seven patients with PM and DM refractory to steroid and/or immunosuppressive agents, researchers combined MMF with intravenous immune globulin (IVIg), which was associated with complete remission.(28) Case series and uncontrolled studies have suggested efficacy for MMF in treating refractory cutaneous DM.(29)(30)
Over the past decade, MMF has gained popularity as a treatment of myositis-ILD. Two small case series suggested efficacy for MMF in treating systemic autoimmune rheumatic disease (SARD)-related ILD (CTD-ILD).(31)(32) In another case series of four patients with DM-associated ILD on prednisone, the addition of MMF resolved dyspnea and normalized pulmonary function tests (PFTs) in three patients after one year of follow up, and improved the diffusing lung capacity (DLCO) in the other patient.(33)
In the largest cohort of SARD-ILD, 125 patients (32 with PM or DM) treated with MMF for a median of 897 days showed significant improvements in forced vital capacity (FVC) at 52, 104 and 156 weeks and DLCO at 52 and 104 weeks.(34) There were trends toward statistically significant FVC% and DLCO% improvement among subgroups with PM or DM at 52, 104 and 156 weeks. MMF use has also been reported for treatment of rapidly progressive ILD in a patient with clinically amyopathic dermatomyositis.(35)
MMF is administered orally starting at 250-500 mg twice daily and increased by 250 to 500 mg increments every one to two weeks until reaching the target dose of 2000-3000 mg/day. A lower dose is recommended in patients with renal insufficiency.
Cyclosporine and Tacrolimus
T-lymphocytes have been suggested as targets in the management of myositis-ILD. One study revealed that infiltrating lymphocytes in IIM-ILD patients included activated CD8+ T-cells.(36) In another study, a decrease in regulatory T-cells in interstitial pneumonitis was noted in rheumatic disease.(37) Tacrolimus and cyclosporine have been used for treating inflammatory myopathy, particularly those with coexisting ILD.(38)(39)(40)(41)(42)(43)(44)
Cyclosporine is a calcineurin inhibitor that inhibits production and release of interleukin 2 and interleukin 2-induced activation of T-lymphocytes. For 14 patients with DM and acute/subacute interstitial pneumonia, combination therapy with prednisolone (1 mg/kg/day) and cyclosporine (4 mg/kg/day) within 12 days from diagnosis improved both PFT parameters and high-resolution computed tomography (HRCT) findings.(40) In another retrospective study of eight anti-Jo-1 positive PM patients with ILD treated with oral cyclosporine, progression on HRCT was not significantly different from those receiving cyclophosphamide.(41) A more recent study of 48 patients with DM-ILD from Asia reported that early cyclosporine treatment was associated with significantly improved survival compared to delayed cyclosporine treatment.(45)
Tacrolimus is a second generation, calcineurin inhibitor that binds to an intracellular protein, FKBP-12, resulting in inhibition of T-cell activation. In a case series of eight IIM patients (six with anti-Jo-1 and two with anti-SRP auto-antibodies; five with ILD), tacrolimus therapy led to an improvement in muscle strength and CK in all patients and improvement in PFT parameters in three of five patients with ILD.(43) The follow-up report on 13 patients with anti-synthetase-associated ILD (12 with anti-Jo-1 and one with anti-PL-12 autoantibodies), who received tacrolimus for an average of 51 months, showed significant improvement in muscle strength, CK and all PFT parameters.(44) In another series of 16 PM patients and 15 DM patients, tacrolimus improved the serum CK and muscle strength two to four months later.(46) In a more recent retrospective study, 49 previously untreated patients with PM or DM-ILD received tacrolimus (dose adjusted to a trough level of 5 to 20 ng/mL) plus conventional therapy or conventional therapy (prednisolone, IV cyclophosphomide and/or cyclosporine) alone.(47) The addition of tacrolimus was associated with significantly longer event-free survival.
Three small series of patients with myositis-ILD demonstrated that tacrolimus was beneficial in those refractory to cyclosporine.(38)(44)(48) Tacrolimus has been used as one of the first-line glucocorticoid-sparing agents(49) but its use is generally reserved for patients with refractory disease due to concerns about its potential side effects and toxicity profile including nephrotoxicity.
Cyclophosphamide
Cyclophosphamide is generally reserved for IIM patients with severe or rapidly progressive ILD, overlapping systemic vasculitis or those refractory to several other second- or third-line agents. The reasons include lack of evidence suggesting efficacy in the primary disease and concern about its serious side effects including an increased risk of malignancy.(50)
Cyclophosphamide use in myositis-ILD has been reported in a few case reports and small case series.(51)(52)(53) Seventeen patients with ILD were treated with monthly intravenous cyclophosphamide (300 to 800 mg/m2 monthly) for at least six months, in addition to daily prednisone.(51) Dyspnea was reduced in eleven patients; six of seven patients who required supplemental oxygen were later able to discontinue its use. Twelve patients showed improvements in forced vital capacity (FVC) of at least 10 percent or demonstrated at least a 10-point reduction in a quantitative HRCT score.
Intravenous Immune Globulin (IVIg)
IVIg is an immunomodulatory agent thought to suppress immune-mediated processes. IVIg demonstrated efficacy in DM in a double-blind, controlled trial in 15 patients with refractory DM.(3) In another open label trial with thirty-five PM patients, IVIg therapy, 70 percent showed significant clinical improvement, and the efficacy was maintained in half the patients three years after stopping IVIg.(54)
An alternative subcutaneous form of IVIg in seven patients (four DM, three PM) was associated with significant CK improvement, muscle strength, quality of life, discontinuation of IS agents and reduction of the maintenance prednisone dose in all patients.(55) Subcutaneous IVIg was administered by a programmable pump and the patient's usual IVIg monthly dose was divided into equal doses given subcutaneously at weekly intervals.
In a more recent. double-blind. placebo-controlled trial in Japan, 26 subjects (16 PM and 10 DM) were randomly assigned to receive either IVIg therapy with polyethylene glycol-treated human IgG or placebo. Statistically significant improvements in the primary endpoint (manual muscle test score) and secondary endpoints (serum CK level and activities of daily living score) were noted in both IVIg and placebo groups.(56)
A few case reports have suggested efficacy for IVIg in the treatment of myositis-ILD.(57)(58) In one report, a patient with amyopathic dermatomyositis-associated ILD, refractory to high-dose glucocorticoids and cyclosporine A, showed a good response to IVIg therapy.(59) The 2012 American Academy of Neurology guidelines support IVIg therapy for refractory DM but note insufficient evidence to support or refute its use in PM.(60)
IVIg is usually administered as infusions of 2 g/kg monthly but the dose or interval can be changed based on the disease severity and treatment responsiveness. A major advantage of IVIg is that it is safe in the setting of active infection and can also be used concomitantly with other IS agents. The high cost of IVIg may influence decisions on its long-term use. Therefore, IVIg is generally reserved for patients with severe dysphagia, refractory primary disease including severe cutaneous features or as a salvage therapy in patients with severe and progressive myositis-ILD resistant to conventional IS therapy.
Rituximab
Rituximab, a B cell depleting agent, is a monoclonal antibody against the CD20 antigen on B lymphocytes. The efficacy of rituximab in refractory IIMs has been reported in several small case reports and case series:(61)(62)(63)(64)(65) a small, open-label, uncontrolled, pilot trial of rituximab therapy (four weekly IV doses) in six treatment-resistant DM patients, showed that rituximab therapy was associated with major clinical improvement in muscle strength and rash in all subjects;(66) another small, open-label trial of rituximab in four patients with refractory PM, demonstrated normalization of muscle strength and significant decline in serum CK levels in all patients;(67) however, in an open-label trial of rituximab in eight patients with DM, cutaneous disease (skin scores based on Dermatomyositis Skin Severity Index) and CK levels did not significantly change from those at baseline and only three patients showed modest improvement in muscle strength(68)
In the largest randomized, double-blind, controlled clinical trial in IIM [the Rituximab in Myositis (RIM) Trial], 195 patients (75 with PM, 72 with DM and 48 with JDM; all refractory to glucocorticoid therapy and at least one IS drug) were randomized to receive two 1-gram rituximab infusions either at baseline or eight weeks later.[1] Entry criteria included fairly significant muscle weakness (not required in the juvenile DM patients) and ??JPY2 additional abnormal consensus CSM for adults and ??JPY3 abnormal CSMs with or without muscle weakness for the pediatric subjects. Glucocorticoid and/or IS use was allowed at study entry. The primary end point was the time to achieve the IMACS definition of improvement (DOI) that was compared between the two groups (rituximab early and rituximab late). The secondary end points included the time to achieve ??JPY20% improvement in muscle strength and the proportions of patients in the early and late rituximab groups achieving the DOI at week eight (the time-point at which half the subjects had received rituximab eight weeks earlier, while the other half received placebo).
Although the early rituximab group demonstrated no faster response to therapy than the group that received rituximab later (thus failing to meet the primary outcome), the DOI was met by 83 percent of this refractory group of IIM patients with a median time to achieving the DOI of 20 weeks. Rituximab therapy was also associated with a significant steroid-sparing effect, as the mean prednisone dose decreased from 20.8 mg at baseline to 14.4 mg daily at the end of the clinical trial. Moreover, patients who initially met the DOI and who were subsequently re-treated with rituximab after a disease flare responded to retreatment with rituximab as well.
Rituximab therapy was generally well tolerated and the most common adverse events were infections. Additional analysis of the RIM Trial data demonstrated that the presence of anti-synthetase and anti-Mi-2 autoantibodies along with the juvenile DM subset, as well as lower disease damage, were strong predictors of clinical improvement and response to rituximab(69) The primary and strongest predictor variable on univariate analysis was the presence of the aforementioned autoantibodies.
The efficacy data of rituximab therapy specific to myositis-ILD is limited to uncontrolled studies.(63)(64) In a recent retrospective study of 50 patients with severe, progressive ILD (10 with IIM-ILD), rituximab therapy was associated with a median improvement in FVC of 6.7% (p<0.01) and stability of the DLCO (0% change; p<0.01) in the 6-12-month period after B cell depletion therapy.(70) Among the SARD-ILD patients included in this study, the best results were observed in patients with myositis-ILD as five of the 10 (50%) patients demonstrated an increase in their FVC >10% and/or an increase in their DLCO >15%, compared to four of 22 (18.2%) patients with other SARD-ILDs (p?=?0.096).
A retrospective Norwegian study identified 24 patients with anti-synthetase syndrome and severe ILD with more than 12 months follow-up (median 52 months) post-rituximab therapy.(71) The median percentage of predicted FVC, FEV1 and DLCO increased by 24%, 22% and 17%, respectively, following rituximab therapy. High-resolution CT (HRCT) findings (assessed and expressed as a percentage of total lung volume involvement) showed a median of 34% reduction in ILD extent following rituximab therapy. Muscle strength also increased post-rituximab and the CK significantly dropped. All subjects demonstrated a decrease in their anti-Jo-1 levels with median drop of 33% (p<0.008).
One limitation of the study was combined therapy with another IS agent, as 10 of the 24 patients also received cyclophosphamide making it difficult to attribute the response to rituximab alone. The best outcome (>30% improvement in all three PFT parameters) was observed in seven patients with a disease duration <12 months and/or an acute onset/exacerbation of ILD. There were seven deaths among the 24 rituximab-treated patients, six with infections (including three with p. jirovecii pneumonia).
In a recent multicenter, open-label, phase II trial, 10 anti-synthetase antibody-positive patients with refractory IIM and ILD (refractory to prednisone and at least two immunosuppressive agents), received 1 g of rituximab at day 0, day 15 and after 6 months.(72) Seven patients improved their muscle strength and the serum CK level decreased from 399 IU/L (range, 48-11,718) to 74.5 IU/L (range, 40-47,857). Rituximab therapy was associated with a significant steroid-sparing effect, as the mean prednisone dose decreased from 52.5 mg/day (range, 10-70) at baseline to 9 mg/d (range, 7-65) along with a concomitant drop in IS therapy. ILD improved in five patients and stabilized in four others.
Another retrospective study tracked anti-Jo1 autoantibody positive patients, of whom 17 received rituximab and 30 patients were treated with conventional IS drugs, following them for a mean of 35 and 84 months, respectively.(73) Sixteen of the 17 receiving rituximab demonstrated a more rapid and marked response. In contrast to conventional IS drugs, response to rituximab was independent of the anti-Ro52 antibody status.
Rituximab is usually administered as two 1-gram doses two weeks apart but the interval may vary and the dosing of rituximab has not been standardized for the treatment of IIM. The decision on additional courses of B cell depletion therapy is generally made on a case-by-case basis. All patients should be screened for hepatitis B prior to therapy and high-risk patients require hepatitis C screening. Patients with a history of recovery from prior hepatitis B infection need to be monitored closely for clinical and laboratory evidence of HBV reactivation during rituximab therapy and for one to two years thereafter. Some suggest periodic monitoring of peripheral B cell flow cytometry to assess for the return of CD20-positive B cells. The most common adverse effects of rituximab include infusion-related reactions, cytopenia and infections.
Anti-tumor Necrosis Factor (anti-TNF) Agents
Anti-TNF agents, etanercept and infliximab, have been used for the treatment of IIM but the outcomes have been mixed and their efficacy in IIM is unclear at this time.
In a series of five patients with DM resistant to glucocorticoid and cytotoxic therapy, etanercept (25 mg subcutaneously twice a week for at least three months) was associated with worsening muscle weakness, elevation of muscle enzyme levels and unchanged DM rash in all patients.(74) After the discontinuation of etanercept, the patients improved with the combination of methotrexate and azathioprine. In contrast, in a more recent randomized, double-blind, controlled trial of etanercept (50 mg subcutaneously weekly for 52 weeks) in 16 patients with DM, etanercept use was associated with a significantly longer median time to treatment failure (358 days vs. 148 days; p= 0.0002) and a significantly lower average prednisone dose after week 24 (1.2 mg/day vs. 29.2 mg/day; p = 0.02).(75) Given the small number of patients in this study and the earlier negative study of etanercept in IIM, further studies are needed to clarify the efficacy of etanercept in IIM.
A few anecdotal reports suggested efficacy for infliximab in IIM.(76)(77)(78) Two patients, however, who initially appeared to respond to infliximab had an exacerbation of their myositis, and resuming infliximab led to anaphylaxis and the development of anti-dsDNA auto-antibodies.(79) A larger retrospective series of eight patients with refractory DM or PM found that infliximab use was associated with improved muscle strength and fatigue but only a partial drop in serum CK levels.(80) And a more recent pilot study of 13 patients with refractory IIM reported that infliximab therapy (four infusions of 5 mg/kg body weight over 14 weeks) was ineffective, with no patient showing improvement in their muscle strength.(81) An unpublished randomized controlled trial of infliximab in IIM also failed to demonstrate efficacy.(82) A multicenter, open-label, controlled trial of infliximab in combination with weekly methotrexate in patients with PM or DM was terminated prematurely because of a low inclusion rate and high dropout due to disease progression and the occurrence of infusion reactions.(83) In general, anti-TNF therapy is not routinely used in IIM because of negative studies as well as recent reports suggesting their potential for inducing PM and DM.(84)(85)(86)(87)
Anecdotally, anti-TNFs may work in some patients with IIM and in particular for the management of inflammatory arthropathy (e.g., anti-synthetase positive patients).
Adrenocorticotropic Hormone Gel
Adrenocorticotropic hormone (ACTH) gel is a long-acting, full-sequence ACTH that includes other pro-opiomelanocortin (POMC) peptides. Melanocortin receptors are widely distributed in peripheral cells and their activation by natural or synthetic ligands is thought to have anti-inflammatory and immunomodulatory effects.(88)
In a recent retrospective case review, five patients with refractory myositis (three DM, two PM) received ACTH gel subcutaneous injections of 80 U (1 mL) twice weekly (four patients) or once weekly (one patient) for 12 weeks. ACTH therapy was associated with improvement in muscle strength and resolution of rash in all patients.(89) All patients tolerated the ACTH therapy well, and no major side effects were reported. Another series testing ACTH therapy noted improvements among three of four patients with refractory IIM. No significant changes in weight, blood pressure or glycemic control were noted.(90) A registry study enrolled 25 patients (nine patients with DM and 16 with PM; 23 had been treated with glucocorticoids previously and six were on concomitant glucocorticoids) and all were started on 80 units of Acthar subcutaneously twice a week. There were no serious adverse events. The registry is designed to collect data on 100 patients with 12 months of follow up and the longer term data are pending.(91)
ACTH gel has been an FDA-approved treatment for PM and DM since 1952 and its approval was retained by the FDA in 2010. Therefore, some rheumatologists are considering ACTH gel in patients with refractory IIM or those who are unable to tolerate glucocorticoids due to side effects. An open label clinical trial was recently completed to evaluate the efficacy and safety of ACTH gel in refractory PM and DM (ClinicalTrials.gov identifier NCT01906372).
Tocilizumab
Since the approval of tocilizumab, an antagonist of the interleukin-6 (IL-6) receptor, for rheumatoid arthritis, there has been growing interest in evaluating its potential efficacy in other systemic autoimmune rheumatic diseases including IIM. Numerous cytokines are over-expressed in the serum of patients with myositis(92)(93) and while IL-1alpha, IL-1beta, TGF beta1-3 and type I interferon signature are the dominant cytokines expressed in myositis muscle tissue, IL-6 has also been implicated.(94) Elevated levels of IL-6 were observed in serum of adult DM and JDM patients(95) and serum IL-6 levels were significantly correlated with disease activity and both the type I interferon gene and chemokine signature.
In the first report of tocilizumab therapy in IIM, two patients with refractory PM demonstrated improvement in serum CK level and MRI of their thigh muscles.(96) There were no adverse events except for a mild elevation of serum low-density lipoprotein (LDL) in one patient. The first patient was a 40 year-old Jo-1 positive male with PM refractory to conventional IS agents, treated with 8?mg/kg tocilizumab monthly resulting in a drop in the serum CK level and prednisolone tapering from 20 to 6?mg/day. The second patient, a 31-year-old Jo-1 positive male with PM, had repeated flares with the need for prednisolone greater then 12.5 mg/day. After tocilizumab IV infusions at 8?mg/kg every four weeks combined with prednisolone 12.5?mg per day and methotrexate, his disease stabilized with CK normalization and resolution of abnormal thigh MRI findings.
Separately, a 32-year-old Japanese patient with CCP positivity and inflammatory arthropathy, along with an overlap of DM and systemic sclerosis (refractory to cyclosporine, IV cyclophosphamide, IVIg, tacrolimus and combination methotrexate and adalimumab), was treated with tocilizumab with resolution of the DM rash and arthritis and gradual improvement in the muscle weakness and CK elevation allowing glucocorticoid tapering.(97)
An investigator-initiated (University of Pittsburgh) multi-center, randomized, double-blind, controlled trial is ongoing to assess the efficacy of tocilizumab in refractory adult PM and DM (clinicaltrials.gov, NCT02043548).
Abatacept
The costimulatory molecules, CD28 and CTLA-4, are up-regulated in the muscle tissue of PM and DM patients.(98)(99) Abatacept, which targets CD80 and CD86 on antigen presenting cells, was used successfully in a patient with refractory PM.(100) A child with severe recalcitrant JDM including ulcerative cutaneous disease and progressive calcinosis also responded well to combination therapy of abatacept and sodium thiosulfate.(101) A case report from Japan indicated that abatacept therapy led to a favorable outcome in an anti-signal recognition particle (anti-SRP)-positive patient with refractory IIM.(102) In a more recent report from Europe, a patient with severe IIM overlapping rheumatoid arthritis, peripheral vasculitis and ILD, who had been refractory to several conventional and biologic therapies, demonstrated a favorable response to abatacept with good control of myositis.(103)
A randomized clinical trial (ARTEMIS), published as an abstract, assessed the efficacy and potential role of abatacept in refractory IIM,(104) Twenty patients (9 DM, 11 PM; 13 female, 7 male) with active disease after treatment with glucocorticoids and one or more immunosuppressive or immunomodulatory agents for three months or longer were randomized to receive either active treatment with intravenous (10 mg/kg) infusions of abatacept or delayed start for three months. The primary endpoint was the number of improved responders, according to the IMACS definition of improvement (DOI), after six months of therapy.
Seventeen patients were included in the analyses and eight (47%) achieved the DOI after six months of active treatment. No differences were noted between DM and PM or between female and male patients. At three months, five (50%) patients in the active treatment arm were defined as responders while only one (14%) patient in the delayed onset arm was a responder. There were three serious non-drug-related adverse events including a hospitalization due to fracture, worsening in muscle weakness and pre-existing basal cell cancer. Among the six patients with repeated muscle biopsies, there was a significant increase in frequency of Foxp3+ cells, suggesting an effect of treatment on cells in muscle tissue. Abatacept also needs to be assessed for the treatment of IIM-ILD.
Sifalimumab
There is growing evidence that type I interferon (IFN alpha/beta)-mediated innate immunity may be involved in the pathogenesis of IIM. A study of 67 patients with DM, PM and other myopathies found that clusters of genes known to be induced by IFN-alpha/beta were overexpressed in DM patients (n=14) compared to controls.(105) Immunohistochemistry for the IFN-alpha/beta inducible protein MxA (a "downstream" effect of IFN) showed dense tissue staining, further implicating Type I interferon inducible genes in the pathogenesis of IIM. A follow-up study from the same investigators showed similar findings of overexpression of type I IFN genes including IFI27, IFI44L, RSAD2, and IFI44 as the most up-regulated genes.(106)
Other researchers observed a striking IFN signature with increased levels of IFN-regulated chemokines in serum samples of DM patients.(107) The IFN signature and IFN-related cytokines both correlated with disease activity. A separate 56-patient study of patients with adult or juvenile DM, noted that the type I IFN gene, chemokine signature and serum levels of IL-6 correlated with each other and with myositis disease activity.(108)
A phase 1b multicenter, randomized, double-blinded, controlled, clinical trial, published in 2014, assessed sifalimumab, an anti-IFNa monoclonal antibody, in PM and DM.(109) Sifalimumab therapy here led to suppression of the IFN signature in peripheral blood and muscle tissue (66% and 47%, respectively) which correlated with clinical improvement, as patients with ??JPY15% improvement in the MMT had greater neutralization of IFN signature in both peripheral blood and muscle than those with <15% improvement. Sifalimumab is, however, not currently being pursued by its commercial developer.
Conclusions
Despite the lack of placebo-controlled trials, systemic glucocorticoids are considered the mainstay of initial therapy for idiopathic PM and DM, as well as SARD-ILD. Glucocorticoid-sparing agents are often given concomitantly, particularly in patients with moderate or severe disease activity. First-line conventional immunosuppressive agents include either methotrexate or azathioprine. The treatment of refractory IIMs can be challenging and other conventional or biologic immunosuppressive or immunomodulatory agents including mycophenolate mofetil, tacrolimus or cyclosporine, rituximab, IVIg or cyclophosphamide can be used alone or in various combinations.
Over the last decade, there have been several small case series and a limited number of clinical trials attempting to investigate the efficacy of novel drugs including newer biologics. Though the data are currently limited, given the biological plausibility and encouraging small case series results, further research is required to assess the role of new therapies such as ACTH gel, tocilizumab (anti-IL6), sifalimumab (anti-IFN?) and abatacept (inhibition of T-cell co-stimulation).