Course Authors
Julie J. Paik, M.D.. M.H.S. and Lisa Christopher-Stine, M.D., M.P.H.
Release Date: 11/15/2016
Upon completion of this Cyberounds®, you should be able to:
Distinguish the different forms of myositis based on pattern of weakness, EMG findings, MRI findings and muscle histopathology;
List the most common myositis-associated and specific antibodies in polymyositis, dermatomyositis, immune mediated necrotizing myopathy and inclusion body myositis;
Recognize that a statin can trigger an immune mediated necrotizing myopathy;
Describe the autoantibodies that can characterize specific subgroups of myositis and reveal a specific clinical phenotype associated with the myositis-specific or associated antibody.
Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of autoimmune diseases, primarily affecting the skeletal muscles, which cause predominant proximal muscle weakness. The four main types of IIM are dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM) and a relative newcomer — immune mediated necrotizing myopathy (IMNM).
The diagnosis of IIM typically begins with a careful assessment of the pattern of weakness, electromyography, MRI of the muscles when available and muscle biopsy, particularly in cases when a diagnostic skin rash indicative of DM is not present. While MRI of the muscles is a relatively newer diagnostic modality, it can help determine not only the presence of muscle edema, but can also help evaluate the pattern of muscle edema and/or fatty replacement. Muscle MRI can identify muscle biopsy sites since muscle inflammation can be patchy. Furthermore, autoantibody testing is used to define the clinical phenotype of a particular subset of IIM and assess prognosis. In addition to muscle weakness, other organ involvement, such as pulmonary, skin, joint, gastrointestinal and cardiac manifestations, can occur in conjunction with the IIMs.
The Bohan and Peter criteria(1)(2) were first used in 1973 to describe the clinical and histopathological differences between the two primary forms, polymyositis and dermatomyositis. While there have been other criteria, such as the European Neuromuscular Conference (ENMC)(3), that have included more up-to-date muscle histopathologic criteria for the diagnosis of polymyositis, dermatomyositis and IMNM, they have not, typically, been accepted for clinical purposes or for clinical trials.
It is important to note that polymyositis has emerged as more of a diagnosis of exclusion. Inclusion body myositis, IMNM or a genetic muscle disease can be misdiagnosed as PM. Thus, it is important to obtain a family history of neuromuscular disease and exposure to myotoxic drugs, as well as carefully examining the patient for facial, extraocular muscle weakness and scapular winging.
Clinical Features of Dermatomyositis and Polymyositis
The combined incidence of dermatomyositis and polymyositis has been estimated to be 2 per 100,000 annually in the general population in the United States.(4) Dermatomyositis has been reported to have a bimodal incidence pattern, more likely to occur during childhood and then again between age 50 and 70.(5) In contrast, polymyositis is rare in childhood. Similar to other autoimmune diseases, it affects females more than males.
Organ Involvement in DM and PM
Skeletal muscle: Muscle weakness is the most common finding in both dermatomyositis and polymyositis. The presentation of muscle weakness is usually subacute, developing over weeks to months. The pattern of the weakness is proximal and can commonly include neck flexor weakness, which is best tested with the patient supine. Patients will report a gradual decline of muscle strength and describe difficulty climbing stairs, getting up from a chair, reaching over head or combing their hair.
Skin: The skin manifestations of dermatomyositis clearly distinguish it from polymyositis. Gottrons sign/papules and heliotrope ("lilac") rash typically on the upper eyelids is pathognomic of dermatomyositis. Bohan and Peter's proposed diagnostic criteria for dermatomyositis include the following: clinical, histopathologic, EMG and laboratory evidence of muscle inflammation, and the presence of Gottron's (papules on the back of the fingers) or heliotrope rash as a fifth criterion to fulfill the diagnosis of dermatomyositis.
Other characteristic rashes include shawl sign on the nape of the neck, shoulders and upper back, V-neck sign, linear extensor erythema over the knees and elbows which can even be confused with eczema. Scalp involvement can mimic psoriasis with extensive pruritis and flaking. "Holster sign" can also be seen bilaterally on the lateral aspects of the thighs. Periungal erythema with dilated nail fold capillaries can easily be missed if not carefully inspected on a skin exam, but may be one of the earliest cutaneous features of DM. In fact, it has been reported that nail fold telangiectaisas occur in 30% to 60% of patients early in the course of the illness.(6) Since skin involvement can precede the diagnosis or may be subtle, a skin biopsy can be helpful to be certain of the diagnosis and will typically show interface dermatitis.
In more severe skin involvement, patients can also develop erythroderma, or total body erythema, and deep ulcerations of the skin.(7) Skin lesions are very photosensitive and a flare or worsening of the skin disease can be precipitated by both UVA and UVB light, making it imperative that patients are reminded to use a minimum of SPF 30-50 daily and protective layering.
While the spectrum of dermatomyositis can range from severe skin to mild skin involvement, there is also a subset of dermatomyositis without any skin involvement [amyopathic dermatomyositis (ADM) and hypomyopathic DM]. ADM is characterized by the presence of cutaneous manifestations of DM for six months or more with normal muscle enzymes and muscle strength.(8) Hypomyopathic DM is characterized by cutaneous findings consistent with DM and the absence of clinical muscle weakness for at least six months after the appearance of classic rash. In contrast to ADM, hypomyopathic DM patients can have subclinical evidence of myositis either based on elevated muscle enzymes, EMG, MRI of the muscles or muscle biopsy. While patients with ADM do not have muscle involvement, other complications of DM may be significant. Indeed, patients with ADM can have severe and rapidly progressive interstitial lung disease that can potentially drive the disease, requiring immediate intervention with cytoxic agents.
Calcinosis cutis occurs in up to 20% of adult cases and up to 70% of juvenile DM.(9) In adults, it generally does not always follow the disease course of the skin and muscle disease. Patients with calcinosis cutis can have well controlled skin and muscle disease with immunosuppression, and continue to have progressive calcinosis. It can also be quite disabling and painful depending on the location of the calcinosis. It can occur superficially and erupt through the skin, typically in the sites of compression, such as elbows and buttocks. Deeper calcinosis can also present in the groin and abdomen. Surgical excision may be needed for debulking of calcinosis cutis in deeper areas.
Lung: Interstitial lung disease (ILD) can be a major complication of both polymyositis and dermatomyositis. The incidence of ILD has been reported between 5% and 46% and can range from subclinical to rapidly progressive and fatal.(10) The reported prevalence of ILD in myositis ranges from 20% to 78% and is associated with increased morbidity and mortality.(11)(12) One study demonstrated that acute or subacute ILD and diagnosis of ADM (versus PM) were associated with more than a four-fold increased risk of death.(13)
Cough and dyspnea are the most frequently reported symptoms in ILD, although some patients may be asymptomatic. Pulmonary function tests (PFTs) are important to obtain at baseline and should be monitored regularly. PFTs will demonstrate a restrictive impairment and show decreased FVC, total lung capacity, functional residual capacity, residual volume, with a normal or elevated FEV1/FVC ratio, and reduced diffusing capacity of lung for carbon monoxide.(14) In addition to PFTs, high-resolution computed tomography (HRCT) should be the standard imaging modality for detecting ILD, as it helps distinguish fibrosis from active inflammation. Bronchoalveloar lavage is not specific for diagnosis of ILD. An open lung biopsy is typically not required for diagnosis because of complications associated with the procedure.
Other lung complications in both PM and DM include diaphragmatic weakness, which can result in hypoventilation. PFTs will typically demonstrate low lung volumes, reduced maximal inspiratory and expiratory pressures, along with increased residual volumes and normal FEV1/FVC ratio.(15)
Autoantibody testing is very important in patients who have myositis, especially in those with early ILD. The most frequently found autoantibody and the strongest predictive marker for ILD is antihistidyl-tRNA synthetase antibody (anti-Jo1). The prevalence of ILD in patients with anti-Jo-1 antibodies is more than 70%.(10)(15) Anti-PM-Scl positive patients can also have predominant ILD, usually seen in overlap with systemic sclerosis. Anti-MDA5 is another autoantibody that is seen in patients with ADM and ILD. Patients who are anti-MDA 5 positive can have rapidly progressive lung disease, classic "kissing papules" on the fingers and cutaneous ulcerations.(16)
Heart: Cardiac involvement in DM and PM was first reported in 1899, and has a wide range of prevalence (6-75%) due to the reports of almost every component of cardiac structure being affected such as pericardium (pericarditis), myocardium (myocarditis, conduction system abnormalities) and endocardium (mitral valve prolapse).(17)(18) Congestive heart failure occurs in 3-25% of patients, leading to death in 10-20% of patients with PM.(19) Myocarditis resulting in heart failure has also been reported specifically in two cases at our institution who were positive for Jo-1 antibody and therefore should also be considered in the work-up of heart failure in those who have antisynthetase syndrome.(19)
Gastrointestinal: Dysphagia to solids and liquids has been reported in 32% to 84% of patients with myositis. Loss of pharyngsophageal muscle tone can lead to nasal speech, hoarseness, nasal regurgitation and aspiration. On examination, tongue weakness, flaccid vocal cords, poor palatal motion and pooling of secretions can be seen.(20)(21)
Diagnostic Approach — Dermatomyositis and Polymyositis
As mentioned previously, since patients with polymyositis tend to be misdiagnosed, it is imperative that a very careful history and strength assessment be made. Polymyositis is largely becoming a diagnosis of exclusion without muscle biopsy evidence of primary inflammation or a concomitant myositis-specific or myositis-associated autoantibody present.
Pattern of muscle weakness : Both PM and DM will present with subacute onset and proximal muscle weakness. There should not be any significant atrophy or distal weakness, unless it has been chronic.
Muscle enzymes: Muscle enzymes, creatine kinase (CK) and aldolase, are typically elevated and are reported to be up to 50X the upper limit of normal.(22)
Electromyography: Electromyography (EMG) will most often demonstrate myopathic units that are both active and chronic.(22) An irritable myopathy (usually with positive sharp waves and fibrillations) can be seen on EMG; however, in one study, irritable EMG findings predicted an inflammatory histology in only 44% of patients.(23)
Muscle biopsy: Perifasicular atrophy, one of the most specific features of dermatomyositis on muscle biopsy, is a key attribute of the muscle histopathology classification by the ENMC.(3) [See Figure 1]. Other features on muscle biopsy of dermatomyositis include perivasicular inflammation, muscle infarcts and capillary necrosis with membrane attack complex (MAC) deposition on vessel walls. Since MAC deposition staining on vessel walls may not be routinely available, we feel that the presence of perifasicular atrophy is sufficient to confirm a diagnosis of DM since it is specific for DM. It is also more common to find the predominance of CD4 cells.

Figure 1.
Dermatomyositis – Muscle Biopsy.
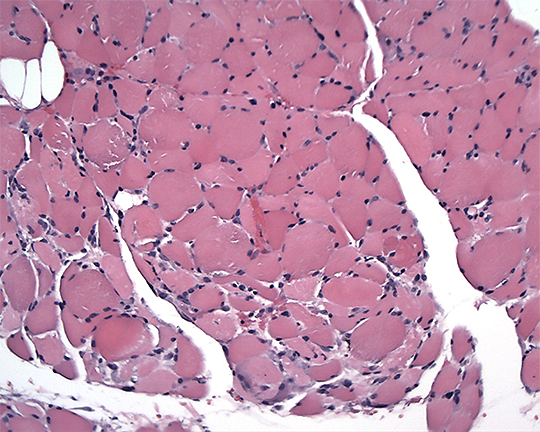
H&E demonstrating perifasicular atrophy.
In PM, we would expect muscle biopsy to demonstrate primary inflammation. Predominance of CD8 positive cytotoxic T cells and activated macrophages are more typical of polymyositis.
MRI of the muscles: Muscle edema is the most common finding on MRI in PM and DM. However, the presence of muscle edema on MRI cannot make a diagnosis of an IIM. While there are extensive reports of specific muscle groups being affected on MRI in neuromuscular diseases, it has not yet been extensively or systematically explored in dermatomyositis or polymyositis. MRI is also helpful in guiding a muscle biopsy to areas of active muscle edema. Thus, while this imaging modality is very useful to determine the presence of muscle edema on T2 weight imaged with fat suppression or short tau inversion recovery, it also gives important insight into the chronicity of the disease by assessing for fatty replacement on MRI.
Autoantibodies: The role of autoantibodies is emerging as an important test to combine with the aforementioned diagnostic studies to make a diagnosis of DM and PM. [See Table 1 and 2.]
Table 1. Anti-synthetase Syndrome: Autoantibodies and Clinical Manifestations.
| Anti-synthetase Autoantibody | Autoantigen | Clinical Manifestations |
|---|---|---|
| Anti-Jo1 | Histidyl t-RNA synthetase | Myositis, ILD, Raynaud's, mechanic's hands, arthritis, +/- DM rash |
| Anti-PL-12 | Alanyl t-RNA synthetase | ILD>myositis, Raynaud's, arthritis, mechanic's hands, +/- DM rash |
| Anti-PL-7 | Threonyl t-RNA synthetase | ILD>myositis, Raynaud's, arthritis, mechanic's hands, +/- DM rash |
| Anti-EJ | Glycl t-RNA synthetase | Myositis, ILD, Raynaud's, mechanic's hands, arthritis, +/- DM rash |
| Anti-OJ | Isoleucyl t-RNA synthetase | Myositis, ILD, Raynaud's, mechanic's hands, arthritis, +/- DM rash |
| Anti-KS | Asparaginyl t-RNA synthetase | Myositis, ILD, Raynaud's, mechanic's hands, arthritis, +/- DM rash |
| Anti-Zo | Phenylalanyl t-RNA synthetase | Myositis, ILD, Raynaud's, mechanic's hands, arthritis, +/- DM rash |
| Anti-Ha | Tyrosyl t-RNA synthetase | Myositis, ILD, Raynaud's, mechanic's hands, arthritis, +/- DM rash |
Table 2. Dermatomyositis Autoantibodies.
| Anti-synthetase Autoantibody | Autoantigen | Clinical Manifestations |
|---|---|---|
| Anti-Mi-2 | Chromatin remodeling enzyme | Prominent skin>muscle disease, with good prognosis |
| Anti-TIF-1 gamma | Transcriptional intermediary factor | Prominent skin disease with diffuse photoerythema has been described. Recently described ovoid palatal patch as a potential distinct cutaneous feature. Skin>muscle |
| Anti-NXP2 | Nuclear matrix protein | Prominent skin disease>muscle, calcinosis |
| Anti-SAE | Small ubiquitin-like modifier activating enzyme | Prominent skin disease>muscle |
| Anti-MDA5 | Melanoma differentiation-associated gene 5 | Palmar papules (can be quite painful), ILD>myositis |
Anti-Jo-1 antibodies, detected in up to 20-30% of patients with myositis, were the first described autoantibodies in myositis.(15)(24) The presence of synthetase antibodies is strongly associated with ILD; and anti-Jo1 is the strongest predictive marker for ILD.
Up to 30% of patients with DM or PM have a constellation of findings called "antisynthetase syndrome" which includes fever, myositis, Raynaud's phenomenon, mechanic's hands, arthritis (typically non-erosive) and ILD.(25)(26)
DM-Specific antibodies include Mi-2, TIF1gamma, MDA5, NXP-2 and SAE.
Anti-MDA5 (melanoma differentiation-associated protein 5) has been emerging as a high risk association for severe ILD and dermatomyositis (particular CADM). In initial studies, it was first described in Japanese cohorts where most patients had CADM and rapidly progressive ILD.(27)(28) The cutaneous findings were not well described in these initial cohorts. Since then, cutaneous findings of ulcers and palmar papules have been linked with ILD and MDA-5 antibodies(29) and are recognized as a "dermato-pulmonary syndrome."
Anti-Mi-2 antibodies are found in 10-30% of patients with IIM and are associated with classical cutaneous features of Gottron's papules, heliotrope rash, shawl sign, V-neck sign, periungal erythema with cuticular hypertrophy and photosensitivity.(30) Anti-Mi2 antibodies are associated with a low risk of ILD and malignancy, and generally confer a favorable treatment response and prognosis.(31)(32)
Anti-TIF1 gamma, or anti-p155/140, are detected in 20-30% of patients with DM. It is strongly associated with malignancy — 70% specificity and 89% sensitivity.(33)(34) It has also been reported that there may be a reduced risk of ILD.(35) Cutaneous features associated with anti-TIF1I? has been reported to include diffuse photoerythema and characteristic 'dusky red face.'(36) An ovoid palatal patch has also recently been described as a potential distinct cutaneous feature of TIF1 gamma positive DM patients who are at high risk of cancer.(37)
Antibodies to NXP-2 occur in approximately 25% of juvenile DM but have also been noted in adult DM.(38) In juvenile DM, it is primarily associated with calcinosis, severe skin disease and joint contractures. There has also been a report of an association with malignancy in adults.(38)
Antibodies targeting SAE, or the small ubiquitin-like modifier activating enzyme (SAE), are seen in 8% of adult DM patients.(39) These antibodies have been associated primarily with skin and muscle involvement, while dysphagia, ILD, arthritis and constitutional symptoms were not reported in an Italian cohort of 183 patients with connective tissue diseases.(40) <
Malignancy: Malignancy has been reported to be in close temporal association with the onset of both dermatomyositis and polymyositis. Breast and ovarian cancer have been most commonly described; however, other cancers such as colon and lung cancer have been reported. Particularly in dermatomyositis, patients who are NXP-2 or TIF1 gamma positive should be considered for evaluation of occult malignancy since they may be at higher risk.(35) Furthermore, in any patient who has refractory disease, a work-up for occult malignancy should be undertaken.
Immune Mediated Necrotizing Myopathy (IMNM)
It is reported than IMNM occurs more frequently than polymyositis and may potentially account for up to 19% of all inflammatory myopathies.(41) Typical clinical features include severe proximal muscle weakness, with highly elevated creatine kinase as well as resistance to conventional immunosuppressive therapy.
It is important to note that, while it is increasingly seen as a separate category of myositis, necrotizing myopathy is a histopathologic diagnosis. Thus, it is not one disease, but can occur with various forms of myositis such as antisynthetase syndrome, scleroderma-associated myopathy, antisignal recognition particle (SRP)-associated myopathy, statin-associated anti-HMG-CoA reductase-positive autoimmune myopathy and statin naA?ve anti-HMG-CoA reductase-positive myopathy.(42) It can also be seen in the context of malignancy and viral infections such as hepatitis C.(43)
Diagnostic Approach of IMNM
Pattern of muscle weakness: Severe proximal muscle weakness usually with more acute onset.
Muscle enzymes: More than 50X upper limit of normal.
Electromyography: Active myopathic unit with irritability (positive sharp waves and fibrillations).
Muscle biopsy: Predominant muscle necrosis with sparse inflammatory infiltrates with concomitant regeneration and degeneration of muscle fibers. [See Figure 2]
Figure 2. Immune-Mediated Necrotizing Myopathy.
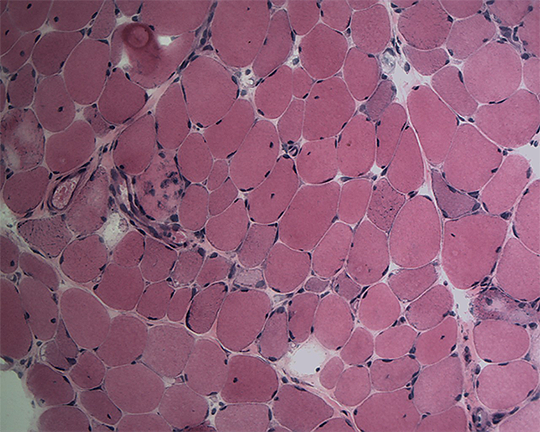
H&E demonstrating necrosis of myofibers without predominant inflammatory infilrates typical of IMNM.
Muscle MRI: Muscle edema with possible early fatty replacement.
Autoantibodies: There are two classic autoantibodies associated with IMNM. [Table 3] The first is anti-SRP. Autoantibodies recognizing one or more constituents of the SRP were first identified in 1986.(44) Although rare (found in approximately 5% of myositis patients), they are associated with particularly severe disease with very high CK levels, dysphagia and severe weakness that may not respond to immunosuppressive therapy.(45)
Table 3. Immune-mediated Necrotizing Myopathy.
| Anti-synthetase Autoantibody | Autoantigen | Clinical Manifestations |
|---|---|---|
| Anti-SRP | Signal recognition particle | Severe, typically recalcitrant muscle disease |
| Anti-HMGCR | HMG-CoA reductase | Statin triggered, can be refractory |
The second autoantibody is anti-HMGCR, which was identified in patients who had statin exposure.(46) This severe form of IMNM progresses even after stopping statins and control may require multiple immunosuppressive treatments. Even without statin exposure, patients can also have anti-HMGCR antibodies, and these statin-naA?ve patients typically fare worse than those who are statin exposed.(47)(48)
Inclusion Body Myositis
Inclusion body myositis is a form of myositis that is insidious, with the onset of first symptoms usually in the fifth to sixth decade of life. It is very important to note that patients with IBM will also have proximal and distal weakness. On average, 12-20 years after onset of symptoms, most patients may require the use of a wheelchair.(49) There can also be mild facial muscle weakness.
Patients with IBM will typically not have any skin or lung involvement, such as ILD, develop over the course of their disease. Malignancy is also usually not associated with IBM. However, recently, there was a report that there may be link with T cell large granular lymphocytic leukemia.(50)
There are multiple classification criteria that have been proposed for the diagnosis of IBM due to the high rate of misdiagnosis. Recently, Lloyd and colleagues performed an unbiased data-directed analysis of 20 features to develop better-performing criteria. They found that the presence of all three of the following had 90% sensitivity and 96% specificity in diagnosing IBM: (1) finger flexion weakness; (2) endomysial inflammation; and (3) either invasion of non-necrotic muscle fiber or rimmed vacuoles.(51)
Diagnostic Approach of IBM
Pattern of weakness: Proximal and distal, important to assess for finger flexor weakness. Quadriceps and forearm atrophy are prominent. Muscle involvement is commonly asymmetric; deltoids and hamstring muscles are relatively spared until the disease is more advanced.
Electromyography: Both myopathic and neuropathic changes can be seen, or a mixed picture, with large and long motor unit potentials that can be misinterpreted as representing a chronic neurogenic process.(52)(53)
Muscle biopsy: Rimmed vacuoles with endomysial inflammation. [See Figures 3 and 4]
Figure 3. IBM.
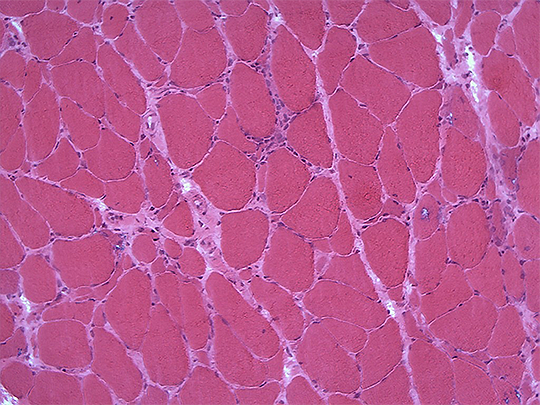
H&E demonstrating endomysial inflammation and rimmed vacuoles.
Figure 4. IBM.
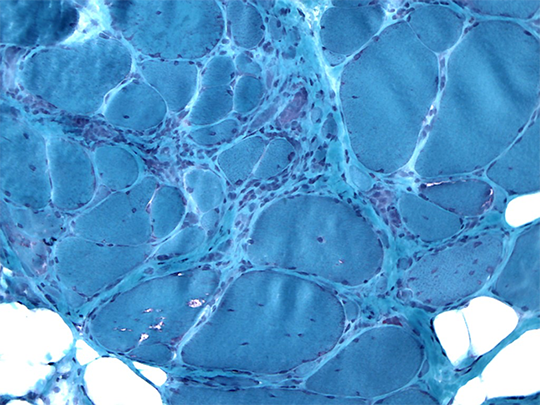
Gimori Trichrome (GT) stain demonstrating red rimmed vacuoles on high power.
Muscle MRI: Muscle MRI demonstrates muscle edema and fatty replacement in the anterior compartment (in the quadriceps).
Autoantibodies: Cytosolic 5'-nucleotidase 1A (NT5C1a) has been noted in a majority of patients with IBM.(54)(55) An enzyme-linked immunosorbent assay testing for IgG, IgM and IgA anti-NT5C1a autoantibodies has been developed that has 76% sensitivity and 96% specificity for identifying patients with IBM.(56) Subsequent reports show anti-NT5C1A may not be entirely specific; while it was found in 61% (71 of 117 patients) of patients with IBM, it was also seen in 5% (2 of 42 patients) with polymyositis, 5% (2 of 42) of healthy volunteers, 15% (24 of 159 patients) with DM and 23% (10 of 44 patients) with Sjogren's Syndrome.
Summary
Idiopathic inflammatory myopathies (IIM) are heterogeneous and include the following four types of myositis: 1) dermatomyositis, 2) polymyositis, 3) immune mediated necrotizing myopathy and 4) inclusion body myositis. The diagnostic approach to evaluating a patient with myositis involves the following: careful history taking, assessment of pattern of weakness, EMG, muscle MRI, muscle biopsy and autoantibody testing. In cases of dermatomyositis, where a classic rash is obvious, a muscle biopsy is typically not necessary. However, given that polymyositis is now becoming a diagnosis of exclusion, and is commonly mistaken for a necrotizing myopathy or early inclusion body myositis, a muscle biopsy can be quite helpful diagnostically. Autoantibodies are becoming more useful frequently determining specific clinical phenotypes that can dictate prognosis, and have proven to be critical in making decisions about management/and or treatment.
Acknowledgement: Andrea M. Corse, M.D., Director of Neuromuscular Pathology Laboratory at Johns Hopkins Hospital, for all muscle biopsy images.