Course Authors
Sarah Canetta, Ph.D., and Christoph Kellendonk, Ph.D.
Release Date: 05/02/2016
Upon completion of this Cyberounds®, you should be able to:
Describe aspects of cognitive functioning that are impaired in schizophrenia, with a particular focus on prefrontal cortex-dependent domains such as working memory and attentional set-shifting;
Delineate several major elements of the neural circuitry that contribute to working memory and attentional set-shifting under normal conditions and that may be compromised in schizophrenia;
Describe several of the neuromodulatory systems within the prefrontal cortex that contribute to working memory and attentional set-shifting under normal conditions and that may be compromised in schizophrenia;
Discuss several emerging areas of therapeutics to treat cognitive dysfunction in schizophrenia.
Schizophrenia is a severe mental disorder characterized by three main symptom domains: positive, negative and cognitive. While positive symptoms such as hallucinations and delusions are largely responsive to antipsychotic medication, negative symptoms like anhedonia, and cognitive symptoms such as memory and attentional deficits, constitute a major unmet therapeutic need. Cognitive symptoms, in particular, have been the focus of substantial on-going clinical and preclinical research as cognitive impairment is highly predictive of long-term functional outcome in patients.(1)(2)
Cognitive impairments in schizophrenia are generalized across a number of neuropsychological domains, including attention, working and reference memory, behavioral flexibility, verbal fluency and processing speed.(3) This generality likely reflects abnormalities in a number of different brain regions. However, this Cyberounds® will specifically focus on prefrontal cortex-dependent impairments (working memory and attentional set-shifting) and their underlying molecular mechanisms, as these are core deficits of the disorder.(4)(5)(6)
Working Memory and Attentional Deficits in Schizophrenia
Although cognitive deficits in patients with schizophrenia occur across multiple domains, impairments in working memory and behavioral flexibility are particularly important because they typically affect sixty to seventy percent of patients.(5)(6) Working memory, our ability to hold information on-line and manipulate it to solve a task, is frequently assessed in patients using an 'N-back' task. In this task, participants are shown a series of images and must identify whether the image they are currently viewing is the same or different from an image presented 'N' trials before; in this task, working memory load increases with increasing 'N'. Patients with schizophrenia perform more poorly on this task than controls as working memory demand increases.(4)(7)
Attentional set-shifting, a measure of behavioral flexibility, is frequently assessed with the Wisconsin Card Sorting Task.(8) In this task, participants are presented with a series of cards that contain cues with three distinct dimension — color, number and shape. The participant has to learn which dimension predicts correct matching through trial and error, and in the process develops an attentional 'set' — the ability to attend to a particular aspect of a complex stimulus while ignoring the irrelevant domains. During the 'set shift' portion of the task, the cue dimension that predicts matching is subsequently switched and participants must learn the new rule in part by inhibiting their old associations. Patients with schizophrenia take more trials to learn the correct rule and inhibit the old one.(8)
Working Memory and Attention in Rodents
Working memory and attentional set-shifting can also be measured in rodents. In mice, working memory is frequently assessed with a delayed non-match to sample T-maze task.(9) This task is comprised of three phases: sample, delay and choice. In the sample phase, mice are forced to explore one arm of the T-maze where they obtain a food reward. In the delay phase of the task, the animals must hold the information they received during the sample phase online in order to choose the opposite arm and receive the remaining reward in the choice phase of the task. Working memory load increases as the delay between the sample and the choice phase of the task increases. Therefore, working memory can be assessed by looking at task performance with increasing delays.
In rodents, attentional set-shifting can be assessed using an odor-based discrimination task.(8) Mice are trained to dig in bowls filled with bedding for a buried food reward. During the task, the bedding is defined by both texture and smell. Initially, one dimension of the bedding (texture or smell) predicts the location of the reward and the animal must learn this rule through trial and error. Once the animal has learned this rule, the dimension that predicts the reward is shifted and the number of trials it takes the mouse to correctly acquire this new rule provides a measure of set-shifting performance.
Mouse models of genetic or environmental risk factors frequently show impairments in working memory or set-shifting tasks. For example, mice with the 22q11 mutation that is a risk factor for schizophrenia show impairments in working memory performance in the T-maze task.(10) Similarly, mice overexpressing striatal dopamine D2 receptors, which model a common imaging phenotype in schizophrenia, also show impairments in the acquisition of the T-maze task.(11) Maternal immune activation, which models an environmental risk factor for schizophrenia, produces offspring with deficits in set-shifting.(12) The fact that these rodent risk factor models produce impairments in cognitive behaviors similar to those seen in patients allows a window into the dissection of circuitry and molecular mechanisms underlying these behaviors, as will be discussed later.
Brain Regions Essential for Working Memory and Attentional Set-Shifting
Although higher order cognitive functioning depends on the coordination of many different brain regions, the prefrontal cortex (PFC) is believed to be particularly essential for working memory and set-shifting behaviors. Evidence for PFC involvement in working memory and set-shifting comes from both functional imaging as well as lesion studies. When participants, for example, perform the Wisconsin Card Sorting Task or an N-back task, activity in the PFC increases with increasing working memory load.(13)(14) Furthermore, people with lesions in these structures show impairments on working memory and attentional tasks.(8)(15)(16)(17)

Figure 1.
Brain Regions and Neurotransmitter Systems Contributing to Cognitive Behaviors Relevant to Schizophrenia.
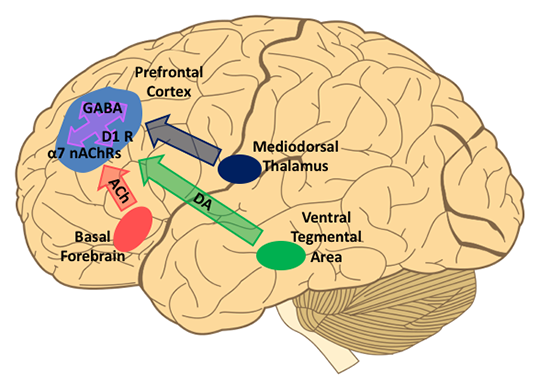
The medio-dorsal nucleus of the thalamus (MD) provides one of the main sources of excitatory input to the PFC. Lesion and imaging studies in humans suggest that the connections between the MD and PFC are essential for cognitive performance, in particular for working memory. MD lesions result in impairments in working memory(18) as well as attentional tasks with high cognitive demand.(19)(20)(21) Imaging studies have also assessed connectivity between the MD and PFC by assessing the temporal dynamics of glucose uptake between these structures. A comparison of the signals generated by [18F]fluorodeoxyglucose positron emission tomography in the MD and PFC of 41 unmedicated patients with schizophrenia and 59 normal comparison subjects performing a verbal learning task revealed a metabolic disconnection between these structures in patients.(22)
Animal studies confirm an essential role for the PFC and MD in working memory and set-shifting. Lesion or pharmacological inactivation of these structures in adult mice impairs working memory(9)(11)(23) and set-shifting performance.(24)(25) In the future, optogenetics — a technique whereby specific cells or brain regions are engineered to express light-responsive ion channels that either hyperpolarize or depolarize the cell in response to focal light administration delivered via an implanted optical probe — may be able to further unravel how these different structures communicate during specific aspects of these cognitive tasks.
Abnormalities in PFC and MD Function in Schizophrenia
As PFC and MD function are involved in normal working memory and set-shifting behavior, there is also considerable evidence that both are compromised in patients with schizophrenia. In functional imaging studies, participants performing an N-back or Wisconsin Card Sorting task show increased PFC activation with increasing working memory load or while engaging cognitive control.(13)(14) In contrast, patients with schizophrenia show reduced task-evoked PFC activation, even when controlling for behavioral performance.(13)(26) Similarly, functional imaging studies have demonstrated that the MD is hypoactive, both at rest and while performing a working memory task.(27) Studies of functional connectivity between the MD and PFC reveal decreased MD-PFC connectivity, both at rest and during performance of cognitive tasks.(22)(28)(29)(30) These functional results lead to the intriguing theory that cognitive deficits in schizophrenia may be partially attributable to impaired functional connectivity between the MD and PFC.
GABAergic, Cholinergic and Dopaminergic Regulation of Working Memory and Attention
In addition to receiving excitatory glutamatergic input from the MD, the PFC is a site of action for a variety of neurotransmitters and neuromodulators, including GABA, dopamine, acetylcholine and serotonin. While there is evidence that all of these systems play a role in working memory and attentional set-shifting under normal circumstances, and that they may be compromised to varying degrees in patients with schizophrenia, this Cyberounds® will focus on the role of prefrontal GABAergic, cholinergic and dopaminergic modulation of PFC function in cognitive processes and in disease.
Prefrontal GABAergic Deficits in Patients with Schizophrenia
GABAergic interneurons can be distinguished into many different subgroups based on histological and electrophysiological properties.(31)(32) Converging functional and histological evidence indicates that interneurons that express the molecular marker parvalbumin (PV) and are capable of firing action potentials at very high rates are particularly affected in the prefrontal cortex of patients with schizophrenia. Several studies have found that levels of GAD67, the enzyme primarily responsible for the production of GABA, are reduced in the cell bodies(33)(34) and presynaptic boutons(35) of PV interneurons.
NMDA receptor activation is also believed to be reduced in prefrontal PV interneurons in patients with schizophrenia, although the evidence for this is mainly indirect.(36) In contrast, interneurons that express a different molecular marker, calretinin, do not appear to be affected.(34)(37) Functionally, these reductions in GAD67 and NMDA receptors are believed to impair the ability of PV interneurons to produce cortical oscillations in the gamma frequency (30-80 Hz), a physiological measure of brain function that is thought to support cognitive processes including working memory and set-shifting, and which are altered in the prefrontal cortex of patients performing cognitive tasks.(38)(39)(40)(41)(42)
Preclinical rodent studies have attempted to causally link alterations in prefrontal PV interneuron function to specific cognitive behaviors. In adults, acute suppression of prefrontal PV interneuron activity using optogenetics led to impairments in several attentional set-shifting tasks analogous to ones impaired in patients with schizophrenia.(12)(43)(44) Similarly, reducing prefrontal PV neurotransmitter release via cell-type specific expression of tetanus toxin impaired performance of several working memory based tasks.(45)
There have also been attempts to mimic specific observed histological changes, such as a reduction in GAD67 and NMDA receptor expression in PV interneurons. Genetic reduction of GAD67 in PV interneurons throughout the brain resulted in several other phenotypes relevant to schizophrenia including sensory-motor gating, increased novelty seeking and impaired fear extinction and social memory, but no reported alterations in working memory; set-shifting was not assessed.(46)(47) In contrast, a reduction in NMDA receptors in hippocampal and cortical PV interneurons led to impairments in spatial working memory and social interaction, in addition to sensory-motor gating and increased anxiety.(48)
These different manipulations affecting PV interneurons result in a number of overlapping and non-overlapping phenotypes. The divergent phenotypes observed may result from differences in the brain regions to which the manipulations were targeted. Cumulatively, however, these studies provide evidence that prefrontal PV interneuron function may be important for cognitive deficits seen in schizophrenia.
Prefrontal Cholinergic Deficits in Normal Cognition and Patients with Schizophrenia
Anecdotally, the high rate of smoking in patients with schizophrenia has been cited as evidence of self-medication to compensate for reduced levels of endogenous nicotinic cholinergic transmission.(49) Indeed, when asked why they smoke, patients reported that it helped them concentrate and improved their focus.(50) This is supported by clinical evidence that nicotine administration improved patient performance in tasks requiring sustained attention, recognition memory and working memory.(49) This was seen in both smokers and non-smokers, suggesting that this effect is not simply counteracting the effects of nicotine withdrawal. Genetically, mutations in the genes that code for nicotinic cholinergic receptors have been repeatedly associated with an increased rate of illness.(49) Furthermore, post mortem analyses demonstrate that patients with schizophrenia have a decreased density of neuronal nicotinic receptors in the brain, especially those of the ?7-containing subtype.(51)(52)
Cholinergic abnormalities have also been proposed to underlie cognitive symptoms in other disorders, like Alzheimer's disease, which also manifest with cognitive impairments such as loss of working memory.(53) Preclinical studies have provided substantial support for the notion that cholinergic modulation of prefrontal function contributes to working memory and attention. Selective destruction of the cholinergic inputs to the prefrontal cortex in rhesus monkeys severely disrupts prefrontal-dependent spatial working memory.(54) Similarly, prefrontal infusion of scopolamine, a muscarinic cholinergic receptor antagonist, into the prefrontal cortex of rats impairs performance in a DNMS working memory task.(55) Additionally, cholinergic lesions in monkeys and rats have been shown to alter attention and recognition memory, although not all of these manipulations were specific to the prefrontal cortex.(55)(56)(57) Together, this work indicates that the cholinergic system may represent an appropriate target for developing pharmaceutical therapies to treat cognitive impairments.
Prefrontal Dopaminergic Deficits in Normal Cognition and Patients with Schizophrenia
Dopaminergic abnormalities are a hallmark of schizophrenia. For example, in the striatum, dopamine release as well as the occupancy of dopamine D2-receptors by dopamine are enhanced.(58)(59) This is believed to contribute to positive symptoms in the disease, as both measures correlate with psychosis and dopamine D2 receptor antagonists are currently the only effective therapeutic treatment for positive symptoms.(60)
The cognitive and negative symptoms, however, remain largely untreated by D2R blockers. A hypodopaminergic state in the prefrontal cortex is hypothesized to contribute to the cognitive impairments, in particular to alterations in working memory.(61) In support of this, amphetamine-induced displacement of a radiolabeled dopamine D2/D3 receptor ligand in the PFC was reduced in patients with schizophrenia relative to healthy controls and the amount of dopamine release was correlated with working memory function in both schizophrenia patients and healthy controls.(62)
In keeping with these findings, preclinical studies in rhesus monkeys demonstrated that prefrontal dopamine depletion led to impaired working memory.(63) Similarly, administration of the dopamine receptor antagonist, haloperidol, impaired working memory.(64) These impairments could be reversed via infusion of a D1 receptor agonist.(64)(65)(66)(67) However, too high a concentration of D1 receptor agonist also impaired working memory, leading to the theory that an inverted 'U' most accurately accounts for the relationship between dopamine D1 receptor stimulation in the prefrontal cortex and optimal levels of cognitive functioning.(66)(68) Unfortunately, attempts to assay prefrontal dopamine D1 receptor binding in the prefrontal cortex in patients with schizophrenia have been extremely equivocal,(69)(70)(71)(72) potentially due to poor specificity of currently available D1-specific radioligands.
Emerging Therapeutics to Treat Cognitive Deficits
Prefrontal PV GABAergic Interneurons
Given the evidence that prefrontal PV GABAergic interneurons play a role in modulating cognitive processes such as attention and working memory, and that their function is compromised in schizophrenia, considerable effort has been put towards developing pharmaceutical agents that target and enhance PV GABAergic interneuron function. NMDA receptor hypofunction on PV interneurons has been hypothesized to be a driving force for PV GABAergic dysfunction.(73) Consequently, co-factors like glycine or D-cycloserine that potentiate NMDA receptor-dependent glutamatergic transmission have been proposed as potential therapeutic targets. In fact, a recent clinical trial found that adjunct administration of D-serine improved negative symptoms in a high risk population.(74) In contrast, adjunct administration of a glycine transporter antagonist recently failed in a Phase III clinical trial.(73)
Oxidative stress has been proposed to be upstream of NMDA receptor hypofunction in PV interneurons.(73)(75) Antioxidant therapy has also, therefore, been proposed as a mechanism to counter NMDA receptor hypofunction. N-acetyl-cysteine (NAC), an antioxidant, has been tested as an adjunct therapy for schizophrenia after it was found to reverse PV interneuron deficits and associated cognitive behavior abnormalities in an animal model of disease.(76) NAC is well-tolerated, has a low side-effect profile and has showed promise as an adjunct therapy in early studies. However, it appears from the preclinical studies that boosting NMDA receptor function or giving antioxidant therapy are most efficacious when given during development. Thus attention to the age and treatment status of the patients to which these drugs are administered will be important for potentially maximizing their utility.
PV interneurons are thought to influence working memory by enhancing prefrontal gamma oscillations. As working memory task-evoked prefrontal gamma oscillations have been shown to be impaired in patients with schizophrenia, another strategy to improve cognitive functioning has been to try to enhance gamma oscillations by non-pharmacological means, such as transcranial magnetic stimulation (TMS).(77) The effect of one session of active or placebo sham 20 Hz TMS stimulation to the prefrontal cortex during a working memory task on task performance was evaluated in healthy subjects or patients with schizophrenia using a double-blind, randomized placebo-controlled trial. TMS increased the magnitude of gamma frequency oscillations elicited during the working memory task in healthy controls and minimized the elevated baseline gamma oscillations seen in patients but did not affect cognitive performance.(78) However, in a subsequent 4-week, randomized, double-blind, placebo-controlled pilot study, where 27 patients performed the N-back task before and after either active or placebo sham TMS, one month of active prefrontal TMS stimulation was sufficient to improve patients' performance on the N-back task to the level of healthy controls.(79)
It remains to be seen whether the changes in working memory seen in response to this stimulation paradigm will be sustained. A recent preclinical study found that a single session of optogenetic stimulation of prefrontal PV interneurons at 40 Hz normalized gamma oscillations and attentional set-shifting performance in a mouse model of cortical interneuron dysfunction for several days following the intervention.(43) This result raises the hopes that the effects of TMS may result in sustained normalization of oscillatory activity and working memory or set-shifting behavior.
Prefrontal Cholinergic Modulation
Modulation of cholinergic tone has been proposed as a method to improve working memory and attentional impairments in patients with schizophrenia. In particular, considerable interest has focused on modulation of ?7-containing nicotinic cholinergic receptors, as several lines of genetic and histological evidence specifically implicate this class of receptors in disease.(49) ABT-126, a selective ?7 nicotinic acetylcholine receptor partial agonist, has been tried as a once-daily adjunctive treatment for cognitive impairments in schizophrenia. The results of a recent randomized, placebo-controlled trial found that 12 weeks of once-daily 10 or 25 mg of ABT-126 led to an improvement in the MATRICS Consensus Cognitive Battery (MCCB) composite score compared to placebo that fell just short of significance in both dosage groups.(80) It appears that this effect was dependent on smoking status, as non-smokers benefited from a significant or nearly significant improvement in MCCB composite score at both doses (p=0.007 for 25 mg and p=0.053 for 10 mg), while smokers' MCCB score did not change. Within the non-smoker group, significant improvements were seen on three of the seven MCCB domains — verbal learning, working memory and attention. Promisingly, these effects were seen even with less than perfect drug adherence over the course of the trial. Further trials will be needed to confirm the efficacy of ABT-126 for improving cognitive functioning in patients with schizophrenia.
Prefrontal Dopaminergic Receptors
As described above, preclinical work from non-human primates and rodents suggests that activation of the prefrontal dopamine D1 receptor, within a certain range of therapeutic efficacy, should improve working memory. However, targeting the dopamine D1 receptor with selective agonists in humans has been hampered by the difficulty in developing highly selective and orally available agents while minimizing the frequency of side effects (most commonly hypotension).(61) A full D1 receptor agonist, dihydrexidine (DAR-0100), appeared promising when given sub-cutaneously based on evidence that it could increase prefrontal brain perfusion at doses that had a negligible side-effect profile.(81)(82) The effects of this drug on working memory and other cognitive functions were minimal.(82)
Recently, the active enantiomer of dihydrexidine (DAR-0100A), which was found to be more potent that DAR-0100 but with equivalent functional activity, was tested as a cognitive therapeutic agent in a proof of concept study. At the highest tolerated dose (15 mg of DAR-0100A administered over 30 minutes), DAR-0100A was found to have essentially negligible D1 receptor occupancy.(83) But because similar low doses of the drug had produced pro-cognitive effects in non-human primates, the effects of either 0.5 mg or 15 mg of DAR-0100A given intermittently over three weeks on working memory and cognitive functioning were examined. Unfortunately, neither dose of DAR-0100A significantly affected any of the cognitive functions examined, although trends were seen for improvement in the CogState Schizophrenia Battery in the high dose group, as well as improvement in attention on the MATRICS battery in both treatment groups83. A separate study found that three 15 mg doses of DAR-0100A was sufficient to improve cognitive performance in the Paced Auditory Serial Audition Task (PASAT) and on the 2-Back to 0-back ratio in patients with schizotypal personality disorder.(84)
Despite these mixed results with the currently available D1 receptor agonists, this remains an active field of drug development with researchers hoping to develop more selective compounds, as well as improved routes of administration, that will help to minimize off-target side effects. Furthermore, it will be important to develop compounds with a prolonged half-life or a slow-release mechanism, as the half-life for the dihydrexidine compounds administered in these studies is predicted to be only on the order of thirty minutes.
Conclusions
This Cyberounds® has focused primarily on the role of the prefrontal cortex in cognitive processes relevant to schizophrenia. In particular, we have discussed emerging evidence that connections from the medio-dorsal thalamus to the prefrontal cortex are critical for working memory and attentional processes, and are likely impaired in patients with schizophrenia. Furthermore, we have discussed evidence that GABAergic, cholinergic and dopaminergic modulation of the prefrontal cortex is important for normal cognitive function, and that these neurochemical systems may serve as appropriate targets for the development of new therapeutics. Although not discussed here, other brain regions, including the hippocampus, also contribute to cognitive processes relevant to schizophrenia.