Course Authors
Nicholas A. Tritos, M.D., D.Sc.
Release Date: 01/13/2015
Upon completion of this Cyberounds®, you should be able to:
Discuss the role of pituitary surgery in patients with acromegaly
Describe the role of current and novel medical therapies in acromegaly
Review the role of pituitary radiation therapy in patients with acromegaly.
Acromegaly is a clinical syndrome occurring as a consequence of growth hormone (GH) excess during adult life.(1) In these patients, GH is almost always secreted by a pituitary adenoma (95% of cases). Of the remaining patients, ectopic growth hormone releasing hormone (GHRH) secretion by an extrapituitary tumor, eutopic GHRH secretion by a hypothalamic tumor (including gangliocytoma) or, very rarely, ectopic GH secretion by an extrasellar tumor is the underlying cause of GH excess. Iatrogenic or surreptitious GH administration in excess may also recapitulate the clinical syndrome of acromegaly. Very infrequently, acromegaly may occur in association with familial or hereditary syndromes, including multiple endocrine neoplasia 1 (MEN 1), familial isolated pituitary adenoma (FIPA), Carney complex or the McCune-Albright syndrome.
The clinical symptoms, signs and complications associated with acromegaly are outlined in Table 1.(2)

Table 1. Symptoms, Signs and Complications Associated with Acromegaly.
| Symptoms | Signs | Complications |
|---|---|---|
| Fatigue | Frontal bossing | Hypertension |
| Headache | Thickened nose and lips | Dyslipidemia |
| Arthralgias |
Prognathism, increased dental spacing and malocclusion | Glucose intolerance and diabetes mellitus |
| Increase in hand and shoe size | Macroglossia | Coronary artery disease and congestive heart failure |
| Excessive sweating | Visual field defects (in patients with tumors impinging on the optic chasm) Goiter Skin tags Enlarged puffy hands and feet |
Stroke Valvular heart disease Arrhythmias Sleep apnea Colon polyps Hypogonadism Vertebral fractures/deformities Osteoarthritis Carpal tunnel syndrome |
The presentation of this disease is indolent, often leading to a substantial delay between the onset of symptoms and diagnosis. Once suspected, the diagnosis of acromegaly can be established based on the presence of elevated age- and gender- adjusted serum insulin-like growth factor 1 (IGF-1) levels and lack of serum GH suppression on oral glucose tolerance testing (OGTT).(1) Magnetic resonance imaging (MRI) examination, using a pituitary protocol, is indicated in patients with biochemical evidence of GH excess in order to identity the underlying cause (Figure 1). Details of the evaluation and caveats associated with the interpretation of diagnostic testing in acromegaly have been presented elsewhere.(1)(3)
The purpose of this Cyberounds® is to discuss current and emerging therapies for patients with acromegaly (caused by a pituitary adenoma). The goals of treatment are to relieve mass effect from a pituitary adenoma and control GH excess, which is important in order to alleviate pertinent symptoms and mitigate the excess morbidity and mortality associated with uncontrolled acromegaly, while preserving or restoring the rest of the patient's pituitary function.
A Patient's Case
A 45-year-old man presented with frequent, loud snoring and daytime somnolence and was referred for evaluation of sleep apnea. He had history of hypertension for five years controlled on lisinopril. On exam, there was frontal bossing, thickened nose and lips, prognathic jaw, macroglossia, skin tags, enlarged hands and feet. A sleep specialist confirmed sleep apnea based on the findings of polysomnography. In addition, the specialist suspected acromegaly and referred the patient to an endocrinologist. Laboratory testing showed: serum IGF-1: 1,025 ng/ml (normal, 90 to 320). After the oral administration of 75 g of glucose, GH levels reached a nadir of 5.6 ng/ml (normal nadir GH <1 ng/ml). The rest of his pituitary function was normal. A pituitary-protocol MRI examination showed a sellar mass, consistent with a macroadenoma (Figure 1).
Figure 1. Coronal T1-weighted, MR Image of the Sella.
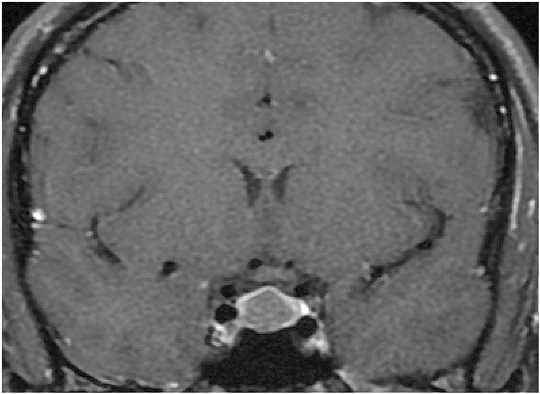
Pituitary macroadenoma in a patient with acromegaly. The tumor appears as a centrally located, hypodense area after gadolinium administration.
Pituitary Surgery
Transsphenoidal pituitary surgery (TSS) remains the mainstay of therapy in most patients with acromegaly.(4)(5). It is the primary therapy for patients with mass effect, including visual field deficits as a result of compression of the optic nerves, chiasm or tracts. In addition, TSS may control GH excess, leading to biochemical remission. Even if complete tumor resection is not possible, TSS may improve the subsequent response to medical therapy by decreasing tumor mass.
Despite technical advances, including the availability of preoperative imaging (MRI and CT), intraoperative use of operating microscope and endoscope, neuronavigation and intraoperative MRI, the procedure remains technically challenging and requires considerable surgical expertise in order to achieve optimal outcomes (4, 5).
Pituitary surgery is very effective in relieving mass effect, including decompression of the optic apparatus. In expert hands, TSS leads to remission of GH excess in 85-90% of patients with microadenomas (defined as tumors <10 mm in greatest diameter) but only in 55-60% of patients with macroadenomas (defined as tumors ??JPY10 mm in greatest diameter).(6) The lower remission rates among patients with macroadenomas occur as a consequence of tumor invasion of the cavernous sinuses, dura or clivus, where complete surgical resection is often difficult to accomplish.
Surgical expertise is also critical to minimize complications, which include epistaxis, tumor bed hemorrhage, cerebrospinal fluid (CSF) rhinorrhea and infection.(4) (5) Transient diabetes insipidus (DI) may occur in approximately 10% of patients but permanent DI is much less frequent (~1%). Transient hyponatremia may also happen during the first several weeks after TSS, necessitating monitoring of serum sodium levels postoperatively. New anterior pituitary hormone deficiencies may appear in 5-10% of patients after TSS. Highly skilled surgeons can perform TSS with very low mortality risk (0-0.5%).
Patients undergoing TSS should have a postoperative MRI examination at 6-12 weeks, as well as thorough reassessment of their pituitary function in order to establish if remission of GH excess has occurred and to detect the possible development of pituitary hormone deficiencies. It should be kept in mind that it may take up to 12 weeks for serum IGF-1 levels to reach a nadir (plateau) postoperatively.(3). A 2-hour OGTT is also advisable postoperatively to confirm the presence of remission. Patients with normal serum IGF-1 levels and suppressed GH levels on OGTT (nadir GH <1 ng/ml or ideally <0.4 ng/ml) are considered in biochemical remission but still need periodic reassessment (at 6 and 12 months and then annually) in order to detect possible recurrence.(3)
Case Follow-up
Our patient underwent TSS by an experienced pituitary neurosurgeon. At 12 weeks postoperatively, his serum IGF-1 level was: 520 ng/ml (normal, 90 to 320). After the oral administration of 75 g of glucose, GH levels reached a nadir of 2.1 ng/ml (normal, nadir GH <1 ng/ml). The rest of his pituitary function remained normal. A pituitary-protocol MRI examination showed postoperative findings without evident tumor.
Current and Emerging Medical Therapies
At present, options for medical therapy in patients with acromegaly include somatostatin receptor agonists (octreotide acetate, octreotide LAR and lanreotide depot), pegvisomant (GH receptor antagonist) and the dopamine agonist, cabergoline, the latter being used "off label" in acromegaly (Table 2).(1)
Table 2. Currently Used Medical Therapies for Acromegaly.
| Name | Mechanism of Action | Dose Range |
|---|---|---|
| Octreotide LAR | Somatostatin receptor agonist (sstr2 and sstr 5) | 10-40 mg every 4 weeks (intramuscularly) |
| Octreotide acetate | Somatostatin receptor agonist (sstr2 and sstr 5) | 100-500 µg every 8 hours (subcutaneously) |
| Lanreotide depot | Somatostatin receptor agonist (sstr2 and sstr 5) | 60-120 mg every 4 weeks (deep subcutaneously) |
| Cabergoline | Dopamine receptor agonist | 0.5-7.0 mg weekly (taken orally in divided doses) |
| Pegvisomant | Growth hormone receptor antagonist | 5-40 mg daily (subcutaneously) |
N.B. Cabergoline use is "off label" in patients with acromegaly.
Medical therapy has been primarily second line therapy in patients with persistent GH excess after TSS. However, primary medical therapy with a somatostatin receptor agonist has also become a viable therapeutic option for patients without mass effect who are unlikely to be cured by surgery (including patients with presumed tumor invasion of the cavernous sinus) or those who decline surgery or are unfit for surgery because of poor health. Medical therapy (generally with a somatostatin receptor agonist) has also been used preoperatively to improve the overall condition of patients with comorbidities that might adversely affect their perioperative risk, such as uncontrolled hypertension or severe sleep apnea/narrow upper airway. It remains unclear if preoperative medical therapy with a somatostatin receptor agonist improves remission rates after TSS.
Somatostatin Receptor Agonists
Short-acting octreotide acetate, octreotide LAR and lanreotide depot are somatostatin receptor agonists that are currently approved by the FDA for use in patients with acromegaly (1). These agents suppress GH secretion by stimulating somatostatin receptor 2 (sstr2) and, to a lesser extent, somatostatin receptor 5 (sstr5). Earlier studies suggested that these medications may control GH excess in up to 60% of patients. However, study subjects were preselected for responsiveness to somatostatin receptor agonists in some of the earlier reports. More recent data suggested that the efficacy of these medications appears to be lower, achieving biochemical control in 20-30% of patients.(7)
Patients treated with long-acting somatostatin receptor agonists (octreotide LAR or lanreotide depot) should have their serum IGF-1 levels monitored 3 months after initiation of therapy, aiming to titrate medication dose to normal serum IGF-1 levels. Somatostatin receptor agonists also lead to some decrease in tumor size (>20%) in the majority of treated patients.
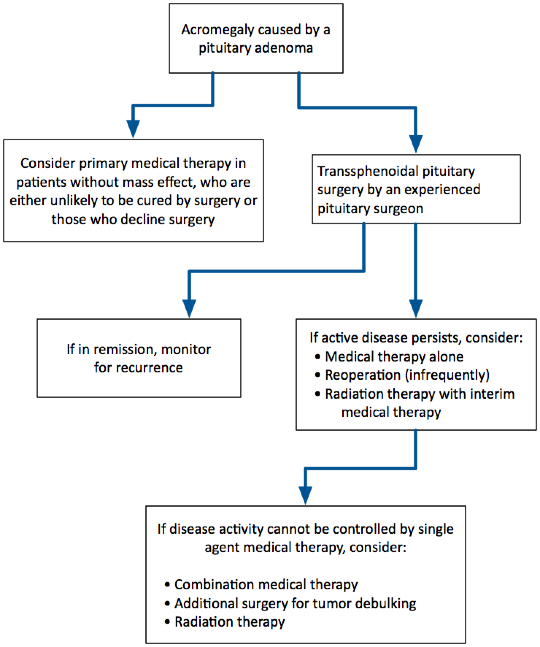
Patients who have a partial biochemical response, but fail to normalize serum IGF-1 levels on somatostatin receptor agonist therapy, may be considered for additional TSS. The rationale for additional pituitary surgery in such cases is based on published data suggesting that pituitary tumor debulking may improve the biochemical response to these medications. Other options for these patients include combination medical therapy (somatostatin receptor agonist plus either pegvisomant or cabergoline) or consideration of radiation therapy (Figure 2).
Figure 2. Proposed Algorithm for the Management of Patients with Acromegaly.
Currently available somatostatin receptor agonists are generally well tolerated. Adverse effects may include gastrointestinal symptoms (bloating, diarrhea, constipation), gallstone formation (usually asymptomatic), hyperglycemia, hypoglycemia, hypothyroidism, vitamin B12 deficiency, bradycardia and hair loss.
Case Follow-up
Our patient was treated with lanreotide depot, which was titrated to a dose of 120 mg every 4 weeks. He tolerated the medication well. His serum IGF-1 became normal: 205 ng/ml (normal, 90 to 320). Sleep apnea resolved on repeat polysomnography, which was performed 12 months later. Screening colonoscopy revealed no colon polyps.
Pegvisomant
Pegvisomant is a GH receptor antagonist, which effectively competes with native GH for binding to the GH receptor without activating downstream signaling pathways.(8) The molecule is pegylated in order to prolong its half-life. Pegvisomant has been approved by the FDA for use in patients with acromegaly.
In pivotal clinical trials, pegvisomant administration led to normalization of serum IGF-1 levels in >90% of study subjects.(8). However, postmarketing observational data have suggested that pegvisomant effectiveness appears to be lower (~65%), possibly reflecting insufficient dose titration.(9) In addition to its use as monotherapy, pegvisomant may be prescribed in combination with either somatostatin receptor agonists or cabergoline and appears to have additive effects in this setting.
It should be kept in mind that GH levels should not be measured during pegvisomant therapy, since pegvisomant does not suppress GH secretion and also cross-reacts with GH in commercially available immunoassays. Pegvisomant does not have direct antiproliferative effects on pituitary adenomas. However, it has been argued that suppression of IGF-1 secretion may remove an anti-apoptotic signal in pituitary adenomas. Observational data suggest that the risk of pituitary adenoma growth in patients treated with pegvisomant monotherapy is low (~3%).
Overall, pegvisomant is well tolerated.(9) Local injection reactions, including lipohypertrophy, have been reported. Rotation of injection sites is recommended to minimize the possibility of this adverse effect. Abnormally elevated liver function tests may also occur, which are generally reversible with a dose decrease or discontinuation of pegvisomant therapy (the latter advised for symptomatic patients or those with markedly abnormal liver enzymes, exceeding three times the upper end of normal). Regular monitoring of liver chemistries is recommended during pegvisomant therapy.
Cabergoline
Cabergoline is a D2 selective dopamine receptor agonist, which is FDA-approved for use in patients with hyperprolactinemia and prolactin-secreting pituitary adenomas. Cabergoline use in patients with acromegaly is "off label."
Cabergoline administration leads to biochemical control of GH excess in 20-30% of patients, when prescribed as mono therapy.(1) Of note, cabergoline doses needed to control GH excess are often higher than those used in hyperprolactinemic patients. Cabergoline is generally well accepted by patients, as the medication is orally active and usually well tolerated. In addition to its use as monotherapy, cabergoline may improve biochemical responses to either somatostatin receptor agonists or pegvisomant, and is particularly effective in patients with mildly elevated serum IGF-1 levels.
Adverse effects associated with cabergoline include nausea, dizziness and, less often, headache, nasal congestion, constipation, vivid dreams or nightmares, anxiety, depression or manifestations of impulsivity. The medication should be avoided in patients with psychosis. Patients with Parkinson's disease receiving high cabergoline doses (>3 mg/day) appear to be at increased risk for cardiac valvulopathy, thought to reflect valvular thickening and fibrosis induced by activation of the serotonin 5HT2B receptor. Hyperprolactinemic patients treated with low-dose cabergoline (0.5-2.0 mg/week) appear to be at low risk of valvulopathy, based on the findings of several, mostly cross-sectional, studies.(1) As some acromegalic patients may be receiving higher cabergoline doses, it is prudent to obtain periodic echocardiograms in this group. The cost effectiveness of echocardiographic monitoring, however, has not been established.
Emerging Medical Therapies
There are several investigational agents being studied as potential therapies for acromegaly, including pasireotide (SOM230), oral octreotide acetate, octreotide implant, CAM-2029, GP02, somatoprim (DG3173) and ATL-1103 (Table 3).(7)(10)(11)(12)(13)(14)
Table 3. Investigational Medical Therapies for Acromegaly.
| Name | Mechanism of Action |
|---|---|
| Pasireotide (SOM230) | Somatostatin receptor agonist (sstr1, sstr2, sstr3 and sstr5) |
| Oral octreotide acetate | Somatostatin receptor agonist (sstr2 and sstr5) |
| Octreotide implant | Somatostatin receptor agonist (sstr2 and sstr5) |
| CAM-2029 | Somatostatin receptor agonist (sstr2 and sstr 5) |
| GP02 | Somatostatin receptor agonist (sstr2 and sstr5) |
| Somatoprim (DG3173) | Somatostatin receptor agonist (sstr2, sstr4 and sstr5) |
| ATL-1103 | Anti-sense oligonucleotide (directed to mRNA encoding the growth hormone receptor) |
Pasireotide is a somatostatin receptor agonist that stimulates multiple receptor isoforms, including sstr1, sstr2, sstr3, and sstr5. The particular ability of pasireotide to stimulate sstr5 is an important pharmacodynamic property that appears to improve its efficacy in achieving biochemical control of disease activity in acromegaly but also provokes a greater risk of inducing hyperglycemia. From earlier studies, we know that short-term pasireotide administration acutely suppressed GH levels in a subset of patients unresponsive to octreotide. A phase II study demonstrated that short-acting pasireotide (administered twice daily) was efficacious in patients with acromegaly.(12)
A sustained-release formulation of pasireotide (pasireotide LAR) was also developed and studied in patients with acromegaly. In a phase III, double-blinded, randomized clinical trial, 358 patients with active acromegaly, based on elevated serum IGF-1 levels and high GH levels measured at random (>5 ng/ml) or on OGTT (nadir GH >1 ng/ml), were enrolled and were randomly allocated to either pasireotide LAR (40 mg intramuscularly every 4 weeks) or octreotide LAR (20 mg intramuscularly every 4 weeks).(7) Of note, 209 patients were treated de novo (that is, in the absence of previous treatment for acromegaly) and 149 patients had persistent disease after undergoing pituitary surgery. At 12 months, biochemical control of acromegaly (normal serum IGF-1 and GH <2.5 ng/ml) was achieved by 31.3% of patients on pasireotide LAR and 19.2% of patients on octreotide LAR, a statistically significant difference (P=0.007). Study subjects in both treatment groups experienced comparable, significant improvements in symptoms and quality of life. Tumor size decreased (by ??JPY20%) in 80.8% of patients on pasireotide LAR and 77.4% patients on octreotide LAR over the 12-month study period.
Gastrointestinal intolerance (diarrhea, bloating, abdominal cramps), gallstone formation, hair loss and headache occurred in both study groups with similar frequency.(7) In contrast, hyperglycemia-related adverse events occurred in 57.3% of patients on pasireotide LAR and 21.7% of patients on octreotide LAR. Data in healthy volunteers have suggested that pasireotide directly inhibits insulin and incretin secretion. Glucose monitoring is advisable in patients receiving pasireotide therapy. If hyperglycemia develops, use of metformin, incretin-based therapies and insulin (as needed) has been recommended.
Available data suggest that pasireotide LAR is more efficacious than earlier somatostatin receptor agonists with regards to achievement of biochemical control in acromegaly. However, use of pasireotide LAR is associated with a higher risk of hyperglycemia.
A formulation of oral octreotide acetate has been developed, which consists of hydrophilic particles containing octreotide acetate in an oily suspension which contains a proprietary transient permeability enhancer.(14) This formulation protects octreotide from gastrointestinal tract digestion and transiently separates the tight junctions between enterocytes, allowing for the passage of octreotide across the intestinal mucosa and eventually into the systemic circulation. Animal data, phase I and II human studies indicated that octreotide can be absorbed and achieve clinically relevant plasma levels after oral administration using this formulation without undue toxicity.
In a published manuscript, a total of 75 healthy volunteers participated in studies in order to investigate pharmacokinetic and pharmacodynamic aspects of oral octreotide acetate.(14) The medication had a significant suppressive effect on GH secretion under both spontaneous and stimulated conditions (the latter occurring as a result of administration of a combination of GH-releasing hormone and arginine). Of note, oral octreotide acetate absorption was significantly decreased in the presence of food or after the administration of proton pump inhibitors. The medication was well tolerated without serious adverse events. Headache, nausea, abdominal pain and diarrhea similarly occurred in some patients after oral octreotide acetate and subcutaneous octreotide administration. There was also transient fecal discoloration noted after oral octreotide acetate administration.
The results of a phase III clinical trial of oral octreotide acetate were presented orally at the annual Endocrine Society Meeting in June 2014 but have not been published at this time. Oral octreotide acetate appears to hold promise as an effective alternative to parenteral somatostatin receptor agonist therapy in some acromegalic patients. However, the need for administration on an empty stomach and possible interactions with other medications might undermine adherence and/or effectiveness of this therapy.
A subcutaneous octreotide implant has also been developed, which delivers the medication gradually over the course of six months.(10)(11) This formulation contains octreotide within a hydrogel capsule, which controls the release of octreotide and is inserted into the subcutaneous tissues in the upper arm under local anesthesia. The results of a phase III study of subcutaneous octreotide implant therapy were published in 2013 (10). In this study, 163 adults with acromegaly, who were selected to be responsive to octreotide LAR therapy, were randomized at a 3:1 ratio to either an octreotide implant (84 mg) or continued on a stable dose of octreotide LAR (10-40 mg every 4 weeks).
Systemic octreotide levels reached a peak between 2-4 weeks after the insertion of octreotide implant. The primary endpoint, which involved normal mean serum IGF-1 and GH <2.5 ng/ml, was achieved in 86% of study subjects on octreotide implant and 84% of subjects on octreotide LAR. In addition, 80 out of 97 subjects (82.5%) indicated that they would prefer the octreotide implant over octreotide LAR therapy (based on greater comfort).
Patients on subcutaneous octreotide implant therapy experienced overall similar adverse events as patients on octreotide LAR. Headache and diarrhea were noted more often in the octreotide implant group, whereas hypertension and cholecystitis were recorded more frequently among patients on octreotide LAR therapy. There was no significant change in fasting glucose or glycosylated hemoglobin. Of note, local site reactions, primarily involving implantation-related complications, occurred in 16 subjects (13.1%) who were allocated octreotide implant therapy.
Currently available data on the efficacy and safety of subcutaneous octreotide implant therapy are encouraging. Additional preclinical studies are in progress.
Several drugs are in earlier stages of clinical development, including CAM-2029, GP02, somatoprim (DG3173) and ATL-1103. Both CAM-2029 and GP02 are novel octreotide formulations in development. The former drug (CAM-2029) is a "liquid crystal" sustained-release formulation, which is administered subcutaneously in prefilled syringes. In phase I studies, administration of CAM-2029 suppressed serum IGF-1 levels in healthy and acromegalic subjects in four weeks. The latter medication (GP02) is a formulation of short-acting octreotide acetate and is delivered by a needle-free injector device through the skin. A phase I study demonstrated that this formulation was bioequivalent to subcutaneous octreotide acetate, based on released, but yet unpublished, data. Healthy volunteers also indicated that they preferred the needle-free device to a subcutaneous syringe/needle. Both medications (CAM-2029 and GP02) are undergoing additional studies to further determine their efficacy and safety in acromegaly.
Somatoprim (DG3173) is a somatostatin receptor agonist that is specific for receptor isoforms sstr2, sstr4 and sstr5 (13). Somatoprim has shown promising findings in preclinical studies, phase I and II clinical studies and is also in clinical development.
Finally, ATL-1103 is an anti-sense oligonucleotide that binds to the mRNA encoding the GH receptor sequence, thus preventing translation and synthesis of GH receptor protein. Administration of ATL-1103 blunted serum IGF-1 and GH binding protein levels in healthy male volunteers by three weeks. Further clinical studies of ATL-1103 are in progress.
Radiation Therapy
Radiation therapy to the sella can be administered as either second-line or, more commonly, third-line therapy in patients with acromegaly who have active disease after undergoing TSS and do not respond, tolerate or agree to medical therapy.(15) Radiation therapy can be administered either as conventional, fractionated, photon-based therapy or as stereotactic radiation therapy, involving use of either a photon beam (Gamma knife, X knife or linear accelerator) or a proton beam.(16) Stereotactic radiation therapy may be administered in a single fraction ("radiosurgery") to patients with tumors that are distant (??JPY4 mm) from the optic apparatus.
Stereotactic radiation therapy decreases exposure of the normal brain structures to low-dose radiation in comparison with conventional radiation therapy. Some studies have suggested that use of somatostatin receptor agonist therapy around the time radiation therapy is administered may decrease the effectiveness of radiation therapy. Accordingly, it is advisable to withdraw medical therapy for several months around the administration of radiation therapy.
Radiation therapy is very effective in achieving tumor control (>90% of cases) but somewhat less effective achieving biochemical control of disease activity (~60% of patients).(15) Drawbacks of radiation therapy include the presence of a substantial lag period (ranging between several months to several years) after its administration until biochemical remission occurs, as well as the frequent occurrence of anterior hypopituitarism (~50% of patients at five years after therapy is administered), necessitating lifelong periodic reassessment of pituitary function.(15)(16) Less common adverse events associated with radiation therapy include the development of optic neuropathy (1-2%), other cranial neuropathies, secondary tumor formation, possibly accelerated atherosclerosis and, rarely, temporal lobe necrosis. Whether stereotactic radiation therapy is associated with a lower risk of some of these adverse events remains to be established.
The availability of several effective medical therapies and the recognition of possible risks of radiation therapy have somewhat tempered the enthusiasm for use of radiation therapy for acromegaly patients, which is most often recommended as a third-line therapy at present (for patients who do not respond adequately to surgery and medical therapy). A proposed flowchart for the management of patients with acromegaly is shown in Figure 2.
Figure 2. Proposed Algorithm for the Management of Patients with Acromegaly.
Summary
Acromegaly is generally caused by a growth hormone-secreting pituitary adenoma and is associated with substantial long-term morbidity and mortality in the absence of adequate treatment, despite its indolent presentation.
A high index of suspicion is needed in order to avoid delayed diagnosis. Once the possibility of acromegaly is considered and confirmed with appropriate testing, transsphenoidal pituitary surgery (TSS) is generally first-line therapy and the therapeutic cornerstone for most patients.
When performed by an experienced pituitary neurosurgeon, TSS leads to disease remission in up to 90% of patients with microadenomas and approximately 60% of patients with macroadenomas. Moreover, pituitary surgery is very effective in decompressing the optic apparatus, leading to vision improvement for the majority of patients with compressive optic neuropathy. Even among patients who do not have biochemical remission of acromegaly postoperatively, tumor debulking, particularly in those patients with tumors invading the cavernous sinuses, dura or clivus, is associated with improved response to subsequent medical therapy.
Medical therapy with a somatostatin receptor agonist, cabergoline or pegvisomant, is generally advised in patients with persistent disease activity postoperatively. Combination medical therapy can be helpful for some patients. In selected cases, primary medical therapy with a somatostatin receptor agonist is an effective treatment option and can be recommended for patients without mass effect who are unlikely to be cured by surgery, or those who decline or are unfit for surgery. Radiation therapy is generally a third-line option for patients who do not respond adequately to pituitary surgery and medical therapy.
Several investigational medical therapies are in development, including pasireotide (SOM230), oral octreotide acetate, octreotide implant, CAM-2029, GP02, somatoprim (DG3173) and ATL-1103, and might offer additional future treatment options.