Course Authors
Mohan N. Viswanathan, M.D.
Release Date: 06/06/2011
Upon completion of this Cyberounds®, you should be able to:
Discuss the extent of the clinical problem of atrial fibrillation
Describe the mechanisms underlying the generation of the cardiac action potential, the ion channels and the factors involved in generating the excitation impulse
Enumerate the leading ideas about the electrophysiological mechanisms underlying the development of atrial fibrillation
Apply the currently available antiarrhythmic medications that are used as rhythm control agents to treat atrial fibrillation, and be able to categorize them via the Vaughan Williams antiarrhythmic classification system
Discuss a few new agents that have just become available or will become available or are under development for atrial fibrillation and the strategies to limit the side effects of the currently available agents.
Cardiac arrhythmias may be seen in individuals with known cardiovascular disease (i.e., coronary artery disease (CAD) or valvular abnormalities) or even in those without known cardiac illnesses. The spectrum of cardiac arrhythmias varies between more self-limited or minimally symptom atic findings such as palpitations, which may be associated with premature supraventricular or ventricular beats that many experience, to the more worrisome ventricular fibrillation, a known cause of sudden cardiac arrest for which emergency cardiac resuscitation is required.(1)
Usually, cardiac arrhythmias are categorized according to the chamber of the heart in which they originate. Supraventricular arrhythmias arise in the atria or in the atrioventricular (AV) nodal structure and usually are not associated with cardiac arrest. Ventricular arrhythmias, on the other hand, originate in the ventricles and are often associated with structural heart disease, especially a cardiomyopathy, whether it is due to CAD or is non-ischemic (idiopathic, not on the basis of prior myocardial infarctions) in origin. Given this is quite an expansive topic to cover, in this Cyberounds® review, we will focus on the most common clinically significant cardiac rhythm disturbance, namely, atrial fibrillation.
Atrial fibrillation (AF) is expected to afflict approximately 2% of the general population by the year 2050 and affects >2 million individuals in the US.(2) When data from the Framingham Heart Study is considered, the lifetime risk of development of AF is on the order of 1 in 4 men and women greater than or equal to the age of 40 years, emphasizing that AF represents not only a challenge to the individual practitioner but a major burden on the health care system in this country and elsewhere. (3)
The use of antiarrhythmic drugs, as well as other agents to control the heart rate when a patient is in atrial fibrillation, can help to curb the untoward symptoms associated with the arrhythmia but their use is limited by their cardiac and noncardiac toxicities. The other factor limiting their use is their marginal efficacy in many patients, necessitating the frequent need to alter the medical regimen in these patients. More recently, invasive approaches have been developed, such as radiofrequency catheter ablation for treatment of AF, which in conjunction with pharmacologic therapy has been an effective strategy to achieve "rhythm control" in individuals suffering from symptomatic atrial fibrillation.(4)
Mechanism of Atrial Fibrillation
Impulse Generation and the Cardiac Action Potential
In order to understand the basis of the various pharmacological treatments for atrial fibrillation, which we will describe later, we must first inspect the cardiac action potential and the various ion channels that are involved at different phases of the action potential. Atrial and ventricular cardiomyocytes are excitable cells and as such they express ion channels on their cell surface. These ion channels display a selectivity to conductance of certain cations, namely sodium (Na+), calcium (Ca++) and potassium (K+). In these excitable cells, the depolarization wavefront engages these ion channels which open and close as a function of voltage across the cell membrane and time. The various ion channel openings and closures that occur across the cell membrane result in the generation of a voltage signal over time, namely the cardiac action potential. Figure 1 shows the action potential as expressed as voltage over time, and displays the slight variation in the contour of the action potential as seen in atrial myocytes and ventricular myocytes. Below, the various currents are shown, and when each is active during the different phases of the action potential. Let’s consider each phase of the action potential and the major currents active in each phase.

Figure 1. Relative Ion Currents In the Atria and Ventricles.
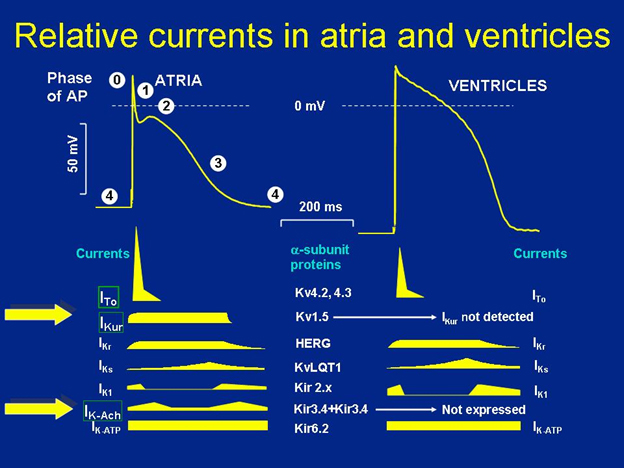
The upper panel of the figure shows the morphology of the cardiac action potential and the subtle differences in the shape of the action potential in the atria versus the ventricles. As shown on the left, the various phases of the action potential are designated with the numbers in the white circles. Phase 4 being the resting membrane potential, Phase 0 being rapid depolarization, and so on. See text for details. The lower panels show the distribution of the various ion channels and currents active throughout the action potential in the atria and ventricles. As can be seen, the ultra-rapid delayed rectifier K+ current, IKur, and the acetylcholine-dependent K+ current, IKAch are not detected in the ventricles (designated with the large yellow arrows). These are atrial-selective ion channels and as such they are being assessed as potential targets for inhibition in order to develop new agents that are atrial-selective and reduce ventricular side effects such as pro-arrhythmia. (Figure courtesy of Dr. Katherine Murray.)
Phase 4 – Resting Membrane Potential
The interior of resting cardiac myocytes is 50-95 millivolts (mV) more negative relative to the outside of the cell due to the distribution of the cations Na+, K+ and Ca++. There is an abundance of open K+ channels at rest, thus the resting membrane potential approximates the equilibrium potential of K+, EK+. Potassium outward current (keeping the inside of the cell negative) through open, inwardly rectifying K+ channels, IK1, primarily determines the resting membrane potential in atrial, ventricular and Purkinje cells (Phase 4 on Figure 1).
Phase 0 – Rapid Depolarization
Once a stimulus is delivered to excitable cells, this evokes an action potential that is characterized by an abrupt change in voltage leading to "depolarization" of these cells, which ultimately "repolarize" and are ready to accept the next stimulus. If the stimulus is sufficient enough to reduce the resting membrane potential to the "threshold voltage" of -70 mV to -65 mV, this is all that is required to produce an action potential, an "all-or-none" response. The upstroke of the cardiac action potential is due to the sudden increase in conductance of the membrane to Na+. As such, the membrane voltage moves toward the equilibrium potential of Na+, ENa+ (Phase 0 on Figure 1).
Phase 1 – Early Rapid Repolarization
After Phase 0, the membrane repolarizes quickly and transiently to below 0 mV (atrial) or nearly 0 mV (ventricle) due to the inactivation of the sodium current, INa, in combination with three other currents. The transient outward K+ current, Ito, is rapidly activated by the depolarized membrane voltage and increases conductance to K+ along its electrochemical gradient. However, it is rapidly inactivated as well, reducing the potassium current before the membrane voltage reaches EK+. The Ca++-activated chloride current, ICl,Ca also contributes to the outward current during Phase 1, promoting repolarization. A third contributor to early repolarization is the Na/Ca exchanger that functions in reverse, leading to an outward Na+ current (Phase 1 on Figure 1).
Phase 2 – Plateau Phase
This phase may last several hundred milliseconds, and the lack of voltage changes during this phase suggests that the conductance of all ions falls to low levels. The plateau voltage is maintained by the competing outward currents of K+ and Cl- ions and inward currents of Ca++ via the open L-type Ca++ channel and Na+ via the Na/Ca exchanger in the forward direction. In Phase 2, K+ conductance reaches almost a standstill despite the large electrochemical gradient for K+ ions at this plateau voltage (Phase 2 on Figure 1).
Phase 3 – Final Rapid Repolarization
During this final phase of the action potential before cells drift back to the resting membrane potential, repolarization occurs rapidly via two currents: a time-dependent inactivation of the L-type Ca++ channel, ICa, L, and activation of the repolarizing K+ currents, the slow and rapid delayed rectifiers, IKs and IKr, and the inwardly rectifying K+ currents, IK1 and IKAch. This results in an increase in the movement of positive ions out of the cell and a return to the resting membrane potential seen in Phase 4 of the action potential. Interestingly, the K+ channel, IKAch, is primarily found in atrial myocytes, and is being investigated as a target for novel atrial-selective antiarrhythmic agents.(5) In fact, in addition to IKAch, the ultra-rapid atrial delayed rectifier K+ current, IKur is also an atrial-selective channel that is active throughout Phases 1-3 of the action potential. Figure 1 displays the various ion currents that are active during the different phases of the action potential, and the large yellow arrows on the left indicate the atrial-selective ion channels that are being targeted for novel antiarrhythmic therapy for AF.
Electrophysiological Basis of Atrial Fibrillation
The definition of AF is an irregular electrical activation of the atria that results in the lack of a distinct P wave on the 12-lead electrocardiogram (ECG). The ventricular response to atrial fibrillation is usually an irregularly irregular heartbeat due to frequent bombardment of the atrioventricular (AV) node by disorganized atrial activity. The three leading hypotheses that underlie the development of AF include: 1) a single ectopic focus or multiple rapidly firing foci with "wave break" of the propagating wavefront; 2) a single "mother rotor" that is associated with fibrillatory conduction; and 3) a multiple wavelet etiology of AF, which is a reentrant and self-perpetuating array of wavefronts that propagate through atrial tissue.(6)(7)
Multiple Wavelet Hypothesis
Preclinical data from animal models have corroborated the multiple wavelet hypothesis; however, there is also data supporting the finding of rapidly firing foci that act as triggers for AF. These foci usually reside in sleeves of myocardial tissue that encircle the pulmonary veins in the left atrium(8)(9) and serve as potential targets of nonpharmacological approaches to treatment of AF via catheter ablation techniques. Given these hypotheses of the arrhythmogenesis of AF, agents that can either reduce the automaticity of individual foci, adversely affect reentry circuits by affecting ion channel conductance, decrease conduction velocity or increase atrial refractory periods can be effective agents to treat AF.
Electrophysiological Reentry
As mentioned, individual foci may trigger AF but what really sustains AF is the development of a substrate that supports reentry. Electrophysiological reentry is the ability of a depolarizing wavefront to "re-enter" a site, forming a continuous circular movement of electrical excitation that repeats itself. It is a function of the path length or wavelength of the impulse as it travels through excitable tissue. This is in turn proportional to the conduction velocity within the tissue and the refractory period:
λ = RP x V
Where λ = wavelength, RP = refractory period and V = conduction velocity. The shorter the wavelength or path length (either due to shorter refractory period or shorter conduction velocity), the more likely reentry can occur and AF, if triggered, can perpetuate itself.(10) Another requirement for reentry is the presence of regions of slow conduction as well as at least two "limbs" of a circuit that have differential refractory periods so as to support more rapid conduction over one limb and conduction block over the other. This allows reverse conduction over the blocked limb and perpetuation of the circuit.
Reentry circuits can be seen around anatomic structures in the heart, especially in the setting of structural heart disease (prior myocardial infarction, prior cardiac surgical scars), or areas of fibrosis termed anatomic reentry. Reentry can also be functional, i.e., an area of tissue is rendered refractory from repetitive wavefronts that create a region of localized conduction block and hence the substrate for reentry. Often, functional reentry can be the underlying mechanism that leads to the early development of AF in normal hearts, potentially beginning with the development of a similar arrhythmia, atrial flutter.(11)
"Atrial Fibrillation Begets Atrial Fibrillation"
To develop the substrate to perpetuate AF, the atria must support a critical number of reentry circuits, and as we’ve seen electrophysiological changes that either decrease the atrial refractory period or lead to conduction slowing will promote reentry. This is the basis of the term coined by Wijffels et al, "atrial fibrillation begets atrial fibrillation."(12) Wijffels’ key experiment showed that AF can be perpetuated for longer periods of time proportional to the duration of time that the tissue is being rapidly paced or "remodeled" to support a shorter refractory period (Figure 2). Electrical remodeling including downregulation of L-type Ca++ channels and the transient outward K+ current, Ito, with an associated increase in the inward rectifier K+ currents, all result in a decrease in atrial refractory period via a shortening of the atrial action potential duration.(13)
Figure 2. Atrial Fibrillation Begets Atrial Fibrillation.
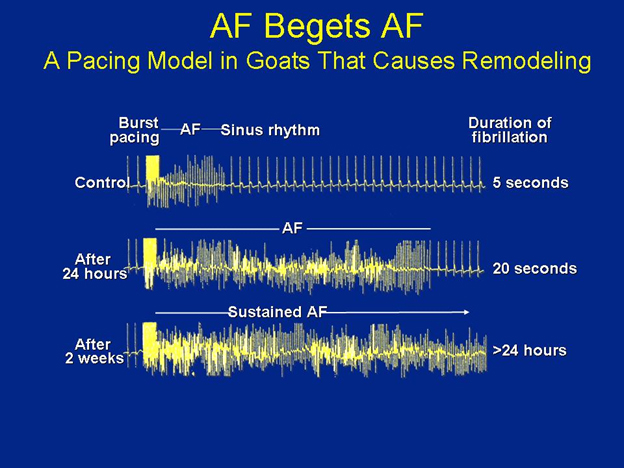
The duration of time that induced atrial fibrillation persists is directly proportional to the duration of time of rapid burst pacing that was applied in this goat model of atrial remodeling and burst pacing of Wijffels and colleagues. The top panel shows that for control animals, there was minimal burst pacing, and the nonsustained AF that remains lasts only a few seconds. In the middle panel, after 24 hours of burst pacing, the induced AF lasted only 20 seconds, while in the last case, after about 2 weeks of rapid burst pacing, the heart may have begun remodeling, and the induced AF lasts greater than 24 hours. (Adapted from: Wijffels, M.C., C.J. Kirchhof, R. Dorland, et al. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995; 92: 1954-68. Reproduced here with permission.)
Contributing Factors to Development and Maintenance of AF
The role of atrial fibrosis has also been recognized as a contributing factor in the development and maintenance of AF. It is commonly observed in ventricular myocardium and may contribute to cardiomyopathies; in the atria the cellular and neurohormonal mechanisms promoting atrial fibrosis have not been fully elucidated.(14) The activation of the renin-angiotensin-aldosterone system has a pro-fibrotic effect and can lead to apoptosis, inflammation and myocardial replacement with fibroblasts.(15)
There is also increasing evidence that inflammation and oxidative stress may be important factors in the pathogenesis of AF.(16) Atrial tissue biopsies from patients with AF have demonstrated histological evidence of oxidative injury, as well as inflammatory infiltrates and fibrotic changes consistent with myocarditis.(17)(18)
Genetic Influences
There are some patients who may have a genetic predisposition to AF, so-called, "familial AF." Recent studies have identified four loci that encode K+ channel subunits (KCNQ1/KVLQT1, the channel responsible for the K+ current, IKs; KCHN2/hERG, the channel responsible for the K+ current, IKr; and two others: KCNE2 and KCNJ2/Kir2.1); mutations in these proteins are associated with a decreased action potential duration resulting in a shorter atrial refractory period, promoting AF.(19)(20)(21) Non-ion channel genes may also contribute to AF development, i.e., mutations in the gap junction protein, connexin-40, have been shown to decrease cell-to-cell excitation coupling, thus reducing conduction velocity and creating regions of slow conduction and the substrate for reentry.(22) See Figure 3 for a schematic diagram of the interplay of ion channels, gap junctions, and pro-fibrotic factors that all may play a role in promoting and perpetuating AF.(23)
Figure 3. Factors Underlying the Development of Atrial Fibrillation.
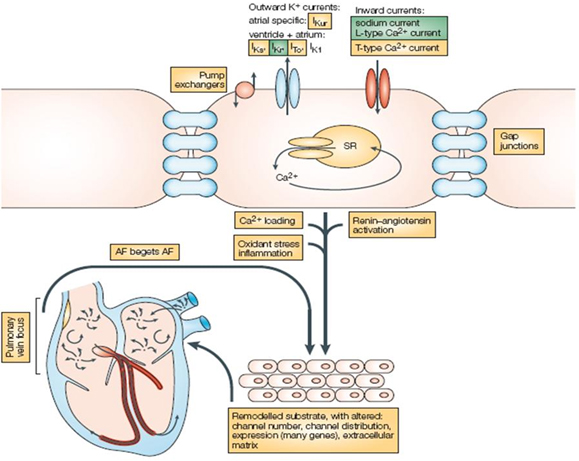
The outward K+ currents, and the inward Na+ and Ca++ currents, as well as gap junctions and ion exchangers, are all shown schematically at the cell surface and may be altered whereupon the stage is set for atrial fibrillation to develop. Other substrate factors such as inciting triggers from a pulmonary vein focus, oxidative stress, inflammatory states, Ca++ loading and continued stimulation by the renin-angiotensin-aldosterone system all may contribute to the development and maintenance of atrial fibrillation. The ultimately remodeled atrium can then perpetuate atrial fibrillation with the alterations listed in the lower portion of the figure. The ion channels or processes that are targeted by currently available anti-arrhythmic agents are highlighted in green, and potentially new targets for drug discovery are highlighted in yellow. (Reprinted by permission from Macmillan Publishers Ltd: Drug therapy for atrial fibrillation: where do we go from here? Nat Rev Drug Discov 2005; 4:899-910.)
Clinical Atrial Fibrillation Management
Categorization of Clinical Atrial Fibrillation
The clinical presentation of AF can be as diverse as the hypotheses that have been developed to explain its electrophysiological basis. Commonly, AF is seen in individuals with structural heart disease, i.e., in those with a cardiomyopathy or valvular heart disease (for example, mitral stenosis). It can occur in the absence of previous heart disease, especially in those less than 60 years of age, this is usually termed "lone atrial fibrillation." The 2006 ACC/AHA guidelines on management of AF have put forth the following nomenclature to better describe the spectrum of AF seen clinically:(24)
|
Termination by an active intervention such as direct-current cardioversion or antiarrhythmic drugs does not change the designation, but persistent AF patients usually need a cardioversion to terminate an episode, as medications may have become ineffective at terminating the arrhythmia. Permanent AF usually signifies AF that has been present often for greater than one year, in which cardioversion has failed or has not been attempted.
Those with lone AF often are able to pinpoint some reproducible triggers of episodes, namely caffeine intake, alcohol, exercise, heavy meals or other vagal influences.(25) They may not have structural heart disease of note, but abnormal atrial pathology such as myocarditis or atrial fibrosis has been identified on biopsies in these individuals.(17) In those patients with permanent AF and valvular disease, ion channel expression may be altered considerably, which may in turn lead to substrate changes that result in an early reversion to AF from sinus rhythm.(26)
Rate vs. Rhythm Control for Atrial Fibrillation
In this Cyberounds® review, we will present current and novel pharmacological options for antiarrhythmic therapy for atrial fibrillation (inherently this is "rhythm control"); however, it is helpful to note a few recent studies that compared the two prevailing strategies of clinical management of atrial fibrillation, rate or rhythm control.
The landmark AFFIRM (Atrial Fibrillation Follow-up Investigation of Rhythm Management) trial randomized 4,060 patients to a rate- versus rhythm-control approach for treatment of atrial fibrillation.(27) The remarkable finding of this largest study to date comparing these strategies was that there was no mortality benefit (primary endpoint) in an intention-to-treat analysis to a rhythm control strategy with antiarrhythmic drugs, primarily amiodarone, over a rate control approach (allowing atrial fibrillation to persist but using medications to maintain the heart rate below 100-110 beats per minute). However, an on-treatment analysis of AFFIRM did show that the presence of sinus rhythm was associated with a 47% reduction in mortality.(28)
The results of AFFIRM were surprising since the prior overarching goal of maintenance of sinus rhythm at all costs was thrown into question, and for select individuals, maintaining the heart rate below ~100 beats per minute and tolerating atrial fibrillation as the predominant rhythm, may be an acceptable strategy. This is similar to the results of the DIAMOND-AF trial, which demonstrated that in patients with reduced left ventricular function, the antiarrhythmic drug dofetilide had no effect on all-cause mortality, but maintenance of sinus rhythm was associated with a significant decrease in mortality (risk ratio 0.44, 95% CI 0.3-0.64, p<0.0001).(29)
It is important to note that in AFFIRM and many of the rate vs. rhythm control studies, patients were excluded from enrollment if they were notably symptomatic in AF, thus these trials tended to include only the minimally symptomatic AF patient, not the typical patient who is seen by cardiologists and cardiac electrophysiologists seeking advanced treatment for their AF.
The AF-CHF (Atrial Fibrillation and Congestive Heart Failure) trial also was recently published. The trial randomized 1,376 patients with a reduced left ventricular ejection fraction (LVEF) of 0.35 or less, symptomatic congestive heart failure (CHF) and documented AF to either a rate or rhythm control approach targeting a primary endpoint of cardiovascular mortality.(4) Once again, the results showed no cardiovascular survival advantage to undertaking a rhythm control strategy in this patient population of AF in the setting of symptomatic CHF and reduced LVEF. Of note, almost 80% of patients were on beta-blocker therapy, 85-90% of patients were adequately anticoagulated, 66% of patients were on digoxin, and 86% of each group were on angiotensin-converting enzyme inhibitor (ACE-I) medications, thus making this trial one of a few in which patients were most appropriately treated for CHF based on the evidence from randomized controlled trials.
Thus, even though there is a lack of survival benefit of pursuing a rhythm control strategy in these trials, maintaining sinus rhythm is still a worthwhile goal, as many patients continue to be intolerant of symptoms associated with atrial fibrillation, and will not accept a rate control approach, especially younger patients.
Current Antiarrhythmic Drug Therapy of AF
The Vaughan Williams classification system has been traditionally used to categorize the currently available antiarrhythmic agents via their various actions on specific ion channels, and alterations in membrane-bound pumps and receptors (See Table 1).(30)
Table 1. The Vaughan Williams Classification of Antiarrhythmic Drugs.
| Drug Class | Agents | Mechanism of Action | Pro- arrhythmic Potential | Normal Heart | Structural Heart Disease |
|---|---|---|---|---|---|
| IA | Procainamide, Quinidine, Disopyramide | Na+ channel inhibition, also K+ channel inhibition, prolongs #APD intermediate onset/offset pharma- cokinetics | QRS widening, ventricular tachy- arrhythmias | Not used as first line | Procainamide: avoided as negative inotrope, Disopyramide: efficacy in hypertrophic cardiomyopathy and AF |
| IB | Lidocaine, Mexiletine (not used in treatment of AF, primarily used for ventricular arrhythmias) | Na+ channel inhibition, shortens APD, rapid onset/offset pharma- cokinetics | Minimal | Often used | Often used |
| IC | Flecainide, Propafenone | Na+ channel inhibition, very minimal increase in APD, slow onset/offset pharmacokinetics | Slowing of cycle length in atrial flutter, 1:1 AV conduction, use- dependent QRS- widening | First line therapy | Contrain-dicated |
| II | β-Adrenergic receptor antagonists (i.e., Metoprolol, Atenolol, Carvedilol) | β1-, β2-receptor inhibition, α-receptor inhibition | Minimal, bradycardia may predispose to *TdP only in setting of QT prolongation | Often used | Often used |
| III | Amiodarone, Sotalol, Dofetilide | Primarily K+ channel inhibition (IKr) leading to prolongation of APD, increased refractoriness Sotalol: IKr and β-receptor inhibition Amiodarone: inhibition of INa, IK, ICaL, β-receptor | Sotalol, Dofetilide: increased risk of TdP Amiodarone: minimal pro- arrhythmia | 2nd and 3rd line therapy | Sotalol, Amiodarone are first line therapy, Dofetilide: second line therapy |
| IV | Ca2+-channel antagonists (i.e., Non-dihydro-pyridine: Diltiazem, Verapamil) | Ca2+ channel inhibition via ICaL, ICaT inhibition | Minimal, bradycardia may predispose to TdP only in setting of QT prolong- ation | Often used | Not routinely used |
Legend:
*TdP: torsades de pointes
#APD: action potential duration
From Vaughan Williams E: Classification of antiarrhythmic actions. Handbook of experimental pharmacology: antiarrhythmic drugs. Vaughan Williams E (Ed.), Springer Verlag, Berlin, Germany (1989):45-67.
Reproduced with permission from: Viswanathan, M.N. and R.L. Page. Pharmacological therapy for atrial fibrillation: current options and new agents. Expert Opin Investig Drugs 2009; 18: 417-31.
Class I antiarrhythmics share a common inhibitory action on the Na+ channel and subclassification of these agents is based on the extent of their effects on repolarization, some via blockade of K+ channels. Class II agents block beta-adrenergic receptor sites at the cell surface and similar to Class IV agents (the non-dihydropyridine Ca++ channel blockers), these drugs are used primarily as rate control agents in AF because of their conduction-slowing properties at the atrioventricular node. The Class III antiarrhythmics prolong repolarization via blockade of K+ channels and include some of the most effective agents, such as sotalol and amiodarone.
The Class I antiarrhythmics exert their action on the action potential during phase 0, rapid depolarization, inhibiting the rapidly opening Na+ channels, and they also have a prolonging effect on action potential duration via their modulation of K+ channels. They have variable binding to open, closed and inactivated Na+ channels; this also serves to subdivide this class into class IA – relatively fast inhibition kinetics, class IB – also fast inhibition kinetics, and class IC – slow onset and offset kinetics.
Quinidine, procainamide and disopyramide are class IA agents (Table 1) that prolong the initial upstroke in phase 0 (Na+ channel inhibition), and increase action potential duration (via K+ channel blockade). They are rarely used clinically due to their side effects -- ventricular proarrhythmia and organ system toxicity. Interestingly, disopyramide has found a use in the setting of hypertrophic obstructive cardiomyopathy (HOCM) as a negative inotropic agent, and is the antiarrhythmic of choice in this patient population with AF.(31)
Class IB agents, lidocaine and mexiletine, are not used for atrial arrhythmias and have their role primarily in the treatment of ventricular arrhythmias.(32) The class IC agents, flecainide and propafenone, as Na+ channel blockers, do slow the upstroke but have no effect on action potential duration (very minimal K+ channel effects). Their slow kinetics of inhibition of the Na+ channel underlies their considerable use dependence (defined as having greater Na+ channel blockade and more slowing of conduction at higher stimulation rates). These agents are contraindicated in patients with structural or ischemic heart disease given the results of the CAST (Cardiac Arrhythmia Suppression Trial) which showed increased cardiac mortality in the post-myocardial infarction patient with flecainide.(33) The precursor of active propafenone has potent beta-blocking effects, and slow metabolizers of propafenone will have increased beta-blockade manifested as bradycardia or significant atrioventricular (AV) conduction slowing.
Class III agents all prolong the action potential duration and, ultimately, the time to repolarization via K+ channel blockade, which is active during phase 3 – final rapid repolarization. Sotalol, dofetilide, amiodarone and now dronedarone are included in this class of antiarrhythmics that can be used in some individuals with structural heart disease. Sotalol is a beta-blocker along with its K+ channel blocking effects via direct inhibition of the K+ current, IKr. Unfortunately, it has pro-arrhythmic effects, namely with prolonged repolarization, the QT interval is prolonged and there is a risk of polymorphic ventricular tachycardia, torsades de pointes (TdP). Dofetilide exerts its K+ channel blockade as a pure IKr blocker, which results in QT prolongation as well, and a risk of TdP is similar to that seen with sotalol. It was shown in the DIAMOND-CHF trial to be effective in patients with CHF and depressed LVEF to convert AF to normal sinus rhythm and was superior to placebo in preventing recurrences of AF and hospitalization for worsening CHF.(34) Dronedarone will be discussed in the section on novel agents even though it has been available for nearly one year.
Amiodarone is classified as a class III agent; however, it displays multiple ion channel inhibitory effects, namely Na+ channel, L-type Ca++ channel, beta-receptor and K+ channel blockade. It is a unique agent in its diverse action on these various ion channels and can lead to an increased action potential duration, prolonged repolarization, and increased refractoriness of atrial and ventricular tissue, i.e., an overall suppressive electrophysiological effect on the heart. It is still the most effective antiarrhythmic available but its use is limited by some of its noncardiac toxicities: thyroid hormone derangements, liver enzyme abnormalities, pulmonary fibrosis from long-term use, and toxicities to the eyes and skin. Meta-analyses have demonstrated that amiodarone has increased efficacy in mitigating AF recurrence compared to sotalol and class I agents.(35)
Novel Agents with Multiple Ion Channel Effects
which has multiple ion channel effects and very rarely is associated with proarrhythmia, is an exception to this rule. On the other hand, it is limited by its other non-cardiac toxicities as listed above. Drug discovery in this area has focused on reducing side effects such as proarrhythmia and the end-organ toxicities seen with amiodarone. To accomplish this, efforts have been taken to develop mixed ion channel blockers that mimic amiodarone in their efficacy. Atrial selectivity has also been a goal of drug design in this area as a way to avoid electrophysiological effects in the ventricle, namely the ventricular proarrhythmia seen with some agents.
Ion channel targets have included the Na+ current, INa, the delayed rectifier K+ currents, IKr, IKs, the ultra-rapid atrial delayed rectifier K+ current, IKur, the transient outward potassium current, Ito, and the acetylcholine (Ach)-dependent K+ current, IKAch. Table 2 displays a number of novel or investigational agents. We will review a few important new agents, but for more in-depth discussions on these agents consider other recent reviews.(36)(37)
Table 2. Selective New and Investigational Antiarrhythmic Agents.
| Agent | Mechanism of Action | Location of effect | Advantages | Disadvantages | Current Status |
|---|---|---|---|---|---|
| Ion channel inhibitors | |||||
| Azimilide | IKr and IKs inhibitor | Atria and ventricles | More effective at higher rates, reduced VT/VF in *ICD pts. | Marginal efficacy | Not approved, and likely will not be pursued |
| Tedisamil | IKr and Ito inhibitor | Atria and ventricles | Multiple K+ blocker, no reverse use- dependence | Extra- cardiac side effects, #TdP risk | Intravenous: Phase III Oral: abandoned |
| Dronedarone | Multiple ion channel inhibitor | Atria and ventricles | Multiple ion channel blockade, less toxicity vs. amiodarone, improved mortality | Increased mortality in ++CHF and $LV dysfunction | Approved and available for use |
| Celivarone (SSR149744C) | Multiple ion channel inhibitor, amiodarone derivative | Atria and ventricles | May be useful in conversion of vagally mediated AF, less side effects than amiodarone | May be less effective than dronedarone, unclear side effect profile | Awaiting dose ranging studies |
| ATI-2042 | Multiple ion channel inhibitor, analogue of amiodarone | Atria and ventricles | Half-life of 7 hours, less side effects vs. amiodarone | Does contain iodine, side effects not known | Phase II efficacy and safety trial pending |
| PM101 | Multiple ion channel inhibitor, amiodarone derivative | Atria and ventricles | Easy intravenous use, rapid onset, water- soluble | May have side effects similar to amiodarone | Phase I studies have been completed |
| JTV-519 | IK1, IKr, INa, ICa inhibition, promotes calstabin-2 binding to RyR2 | Atria and ventricles | Cardio- protective against ischemia, may prevent ||DADs (reduced Ca2+ leak) | Unknown side effects, IKr blockade may precipitate TdP | Only animal studies to date |
| Ranolazine | Late INa, Na+/Ca2+ exchanger, ICaL, IKr, IKs inhibition | Atria and ventricles | Anti- anginal agent, reduced arrhythmias in MERLIN-TIMI 36 | Multiple channel blockade, some QT prolong- ation | Approved for angina, labeling mentions anti- arrhythmic benefit |
| Atrial repolarization-delaying agents | |||||
| Vernakalant (RSD1235) | IKur, Ito, INa, IKr inhibition | Atria >> ventricles | Conversion of AF | Ineffective in AF >7 days' duration, risk of ventricular arrhythmias | Phase III completed; conditional FDA approval for conversion |
| AVE-0118 | IKur, Ito, IKACh inhibition | Atria | Cardioversion of AF, prolonged +AERP in remodeled atria | No clinical studies to date | Phase IIa, develop- ment likely abandoned |
| AZD7009 | IKur, Ito, INa, IKr inhibition | Atria >> ventricles | Atrial selectivity | QT prolongation, TdP risk | Abandoned |
| KCB-328 | IKur inhibition | Atria | Atrial selectivity, terminate atrial flutter | Efficacy unclear | Animal studies only to date |
| Tertiapin-Q | IKACh inhibition | Atria | Atrial selectivity, useful in vagal AF | Unknown side effect profile | Animal studies only to date |
| Other Agents | |||||
| Statins | Anti- inflammatory | Atria and ventricles | Well- tolerated, no pro- arrhythmia | Marginal efficacy | **NIH trial under review |
| Pirfenidone | Anti- fibrotic | Atria >> ventricles | Effects seen in other organ systems | Side effect profile unknown | Animal studies only to date |
| Rotigaptide (ZP-123) |
Gap junction conductance enhancer (connexins) | Atria and ventricles | Novel mechanism, decreased AF vulnerability | Marginal efficacy, unknown side effects | Animal studies only to date |
| Poly- unsaturated fatty acids | Multiple ion channel inhibitor | Atria and ventricles | Non- proarrhythmic, well-tolerated | Marginal efficacy | Clinically available |
Legend: *ICD: implantable cardioverter defibrillator; #TdP: torsades de pointes; $LV: left ventricle; ||DADs: delayed after depolarizations; +AERP: atrial effective refractory period; **NIH: National Institutes of Health; ++CHF: congestive heart failure
Reproduced with permission from: Viswanathan, M.N. and R.L. Page. Pharmacological therapy for atrial fibrillation: current options and new agents. Expert Opin Investig Drugs 2009; 18: 417-31.
Dronedarone
Of all the agents available, dronedarone is the newest member of our armamentarium of antiarrhythmic agents effective against atrial fibrillation. It is an analog of amiodarone but lacks iodine in the chemical structure, and thus was designed to have fewer side effects. Similar to amiodarone, it is classified as a class III agent, though it also has several ion channel effects which include blockade of the ultra-rapid delayed rectifier K+ current, IKur, delayed rectifiers, IKr, IKs, the transient outward current, Ito, and the inward rectifier, IK1, which is mostly active during phase 4 of the action potential.(38) Dronedarone was FDA-approved in mid-2009 and has now been used as an alternative to amiodarone in many patients with atrial fibrillation.
The DAFNE (Dronedarone Atrial Fibrillation Study After Electrical Cardioversion) trial was the first to show benefit of the 800 mg/day dose in preventing recurrences of AF.(39) The next two studies showing benefit of dronedarone were the EURIDIS (European Trial in Atrial Fibrillation or Flutter Patients Receiving Dronedarone for the Maintenance of Sinus Rhythm) and the American-Australian-African counterpart ADONIS trials, which demonstrated that 400 mg twice daily of dronedarone was superior to placebo in preventing AF recurrences and was effective at controlling ventricular rates.(40) Interestingly, dronedarone’s benefit was seen across all the prespecified subgroups, those with or without structural heart disease, CHF and/or hypertension.
It was also noted that dronedarone had a tendency to slow the ventricular rate response in those with AF, i.e., it seemed to be a good rate control agent. To test this idea, the ERATO (Efficacy and Safety of Dronedarone for Control of Ventricular Rate during Atrial Fibrillation) trial was designed and showed that those receiving the drug for 14 days had ventricular rates 12 beats per minute lower than those receiving placebo during a symptom-limited exercise program.(41) Reassuringly, in this study, there were no incidences of TdP in the dronedarone or the placebo arms, suggesting that dronedarone’s proarrhythmic potential is low, making it a good alternative to other class III agents with this untoward side effect.(41)
The ANDROMEDA (Anti-Arrhythmic Trial with Dronedarone in Moderate to Severe Heart Failure Evaluating Morbidity Decrease) trial assessed the effects of dronedarone on all-cause mortality and hospitalizations for heart failure in patients with New York Heart Association functional class III or IV heart failure. This trial was stopped prematurely due to excess mortality in those on active dronedarone treatment versus placebo.(42) This curbed the enthusiasm that was growing in support of dronedarone as an effective novel agent for AF. It seemed that dronedarone was associated with an increase in creatinine, prompting discontinuation of ACE-I treatment in the drug arm, possibly contributing to the excess mortality. It does inhibit renal tubular absorption of creatinine but may not actually negatively impact kidney function.
Even though the results of ANDROMEDA were of concern in terms of dronedarone use in severe heart failure patients, the ATHENA trial, a randomized, placebo-controlled double-blind, parallel arm trial of 4,628 patients, subsequently assessed the efficacy of dronedarone at 400 mg twice daily for the prevention of cardiovascular hospitalization or death from any cause in patients with atrial fibrillation or flutter.(43) Dronedarone significantly reduced the risk of the composite endpoint: first cardiovascular hospitalization or all-cause mortality by 24%, as compared to placebo. Dronedarone did not significantly reduce all-cause mortality alone but there was a statistically significant reduction in cardiovascular deaths compared to placebo.(43) The results of ATHENA taken together with the other trials did serve as the basis for the FDA’s approval of dronedarone.
Novel Atrial Repolarization-Delaying Agents (ARDAs)
Atrial selectivity has been a goal of pharmacological development of new agents for atrial fibrillation given the need to reduce ventricular proarrhythmia, a side effect that plagues many of the currently available agents. The ultra-rapid delayed rectifier K+ current, IKur, is found exclusively in atrial tissue and inhibition of this channel will lead to prolongation of the atrial action potential and the refractory period, thus reducing the predisposition to AF. Given this atrial selectivity, an agent with these properties can potentially minimize ventricular proarrhythmia and QT prolongation.
Vernakalant
The most advanced of the ARDAs is vernakalant (RSD1235), primarily an IKur blocker, which has, however, inhibitory actions on Ito and the Na+ current, INa. In a small Phase II trial of efficacy of the drug, 30 patients with recent onset of AF within 72 hours were given one of two dosing regimens of intravenous vernakalant for conversion to sinus rhythm.(44) The higher dosing regimen was successful at acute conversion (within 30 minutes of the last infusion) in 61% of the patients vs. 11% in the lower dosing regimen, and 5% in placebo. Four other randomized clinical trials of pharmacological cardioversion with vernakalant have been reported with promising results.(45)(46)(47)
The ACT (Atrial Arrhythmia Conversion Trials) trials I, III and IV included subjects with AF lasting between 3 hours and 45 days, whereas ACT II included patients with AF for 3-72 hours within 7 days after coronary artery bypass or valvular surgery. Subjects received a 10-minute intravenous infusion of vernakalant or placebo; if AF persisted, a second slightly lower dose infusion was administered. In ACT I, II and III, vernakalant proved superior to placebo in conversion from AF to sinus rhythm.(45) Notably, the majority converted after the first dose; however, beyond 7 days of AF or atrial flutter, vernakalant was ineffective at conversion. Again, given its atrial selectivity, there was no appreciable QT prolongation with vernakalant. Intravenous vernakalant has been recommended to the FDA for approval for pharmacological conversion of AF.
Substrate-Modifying Agents
Agents without direct action on ion channel conductance can also have important pharmacological action to alter the underlying substrate for AF in the diseased atrium. Often atrial remodeling has occurred over many years promoting the development of AF but a few agents may have promise in altering the substrate to break this cycle of arrhythmogenesis.
The renin-angiotensin-aldosterone system (RAAS) is activated in the setting of atrial stretch associated with atrial enlargement and during AF. Mechanical stretch leads to increased production of angiotensin II and given that the atria have a higher density of angiotensin II receptors than do the ventricles, they are more susceptible to the effects of angiotensin II, which include atrial myocyte hypertrophy, fibroblast proliferation, collagen deposition and myocyte replacement. The result is atrial interstitial fibrosis, leading once again to localized conduction abnormalities, conduction block, and the substrate for reentry is thus set in motion. Angiotensin II can also indirectly affect electrophysiological parameters in the atria via modification of ion channels, leading to an increase in Ca++ influx into the cell, promotion of inflammation in atrial tissue and alteration of cell-to-cell coupling via gap junctions.(36)(48) ACE-Is, as well as angiotensin II receptor blockers (ARBs), can help curb this fibrosis. Both also may have anti-inflammatory actions in the setting of heart failure as well as in AF.(49)
A retrospective analysis of the TRACE (Trandolapril Cardiac Evaluation) trial showed that significantly fewer patients developed AF in the trandolapril group (2.8%) versus those taking placebo (5.3%).(50) Another meta-analysis demonstrated that RAAS inhibitors, ACE-Is and ARBs reduced the relative risk of development of AF by 28% with this effect seen most notably in patients with heart failure or after cardioversion.(51) However, in the largest trial published on the use of renin-angiotensin-aldosterone inhibitors in AF, the GISSI-AF (Gruppo Italiano per lo Studio della Sopravvivenza nell-Infarto Miocardico-Atrial Fibrillation), valsartan, given in addition to standard therapy, which included ACE-Is and amiodarone in some patients, did not decrease the risk of recurrence of AF in patients at risk versus placebo.(52) Obviously, these findings are in stark distinction to the prior data from the meta-analysis mentioned above and, understandably, the role of ARBs in the treatment of AF requires further study.
Similarly, inflammation may be a contributing factor to the substrate perpetuating or triggering AF and this has led to the hypothesis that HMG-CoA reductase inhibitors (statins), given their anti-inflammatory properties, may have a beneficial effect on the substrate for AF. There was a reduced risk of developing AF in patients with stable coronary artery disease and in those who had undergone coronary bypass graft surgery who did receive statin therapy.(53)(54)(55) The ARMYDA-3 (Atorvastatin for Reduction of Myocardial Dysrhythmia after Cardiac Surgery- 3) trial showed a reduction in the incidence of postoperative AF after bypass surgery in patients receiving atorvastatin.(56) The anti-inflammatory effect of statins, their main non-lipid lowering effect, is thought to have a role in curbing the pathogenesis of AF and multicenter trials to assess the benefit of statin use in AF are being planned. Some other substrate-modifying agents that may have a beneficial role in treatment of AF are listed in Table 2.
Conclusions
Atrial fibrillation will continue to be a major cause of cardiovascular morbidity. Despite the increase in our awareness of AF and our understanding of the electrophysiological properties of the atria that predispose to the development of AF, our therapeutic options often fall short. The Achilles heel of the currently available medications for treatment of AF continues to be their marginal efficacy, their tendency to cause ventricular proarrhythmic effects and their cardiac and non-cardiac toxicities. These limitations of currently available agents have fueled a real clinical need to develop new medications that take advantage of some beneficial effects of current drugs, e.g., the multiple ion channel effects of amiodarone, but limit their non-cardiac toxicities.
Our understanding of the importance of the electrophysiological substrate in the perpetuation of AF has highlighted the development and maintenance of reentry as a key factor in the pathogenesis of AF. As we have seen, a shortening of the atrial action potential duration leading to an overall decreased atrial refractory period, along with slowing of conduction velocities, both seem to provide the setting for the development of AF.
By potentially inhibiting certain ion channel effects, and taking advantage of ion channels that are selectively expressed in the atrium versus the ventricle, new medications are being developed that may be able to reverse these electrophysiological changes and restore order to the atria. Furthermore, some new agents may be able to alter the underlying substrate to disallow the development of the electrophysiological changes in the first place.
The challenge in treatment of AF lies in its inherent complexity and despite the development of non-pharmacological treatments of AF (catheter ablation), drug therapy for AF will remain a mainstay of treatment for many individuals. The key will be the tailoring of therapy to the individual patient and making prudent use of the existing agents and new agents as they are developed and become clinically available.