Course Authors
Istvan Merchenthaler, M.D., Ph.D., D.Sc. and Laszlo Prokai, Ph.D., D.Sc.
Release Date: 06/07/2010
Upon completion of this Cyberounds®, you should be able to:
Describe the mechanisms by which estrogen exerts neuroprotective effects
List the efforts to develop a safe estrogen therapy without side effects in the uterus and breast
Discuss concepts to enable safe, central nervous system-selective estrogen therapy.
Following the publication of the book “Feminine Forever” by Robert Wilson in 1966, the use of hormone replacement therapy (HRT or HT) became popular. In the 1980s, numerous observational studies added further support to the concept that HRT would prevent not only hot flashes and osteoporosis but also heart disease, urinary incontinence and dementia, and would thus improve the quality of life.
However, in 1998, the Heart and Estrogen/Progestin Replacement Study (HERS) concluded that the combination of estrogen and progestin increased the rate of cardiovascular disease in the first year. Long-term follow-up showed that there was no overall benefit — indeed the opposite was true — HRT increased the early risk of heart attacks in women with diseased arteries. Then, in 2002, the largest randomized clinical trial of HRT ever conducted, the Women's Health Initiative (WHI) trial and the WHI Memory Study (WHIMS) concluded that the overall risks outweigh the benefits associated with HRT.(1)(2)(3)
The Impact of the Women's Health Initiative (WHI) Trial
The HERS and WHI/WHIMS studies utilized conjugated equine estrogens [Premarin®] plus a synthetic progestin (medroxyprogesterone acetate [MPA]) not the endogenous 17β-estradiol or progesterone the human ovaries produce. Although the estrogen alone arm (ET) of the WHI trial continued, it was also stopped earlier than planned because of increased risk of stroke and no benefit for the prevention of heart disease. ET, similar to the combination therapy (HRT or HT), decreased the risk of hip fracture and colorectal cancer and prevented hot flashes. However, unlike HT, ET had no effect on risk of breast cancer.
The overall conclusion of these studies was that hormone therapy, although preventing hot flashes and hip fracture, increases the possibility of stroke and does not prevent cardiovascular disease. Moreover, the data of the WHIMS trial concluded that ET and HT might be associated with dementia.
In spite of the turmoil the WHI and WHIMS created, there is a growing consensus that the results of these trials should be interpreted within the boundaries determined by the population included, drugs of choice, doses and route of administration, age at which hormone exposure occurs or timing of ET or HT initiation in relation to the onset of the menopause “critical period or window”. At the same time, the WHI and WHIMS studies have also provided the impetus to the scientific community to challenge and reconcile the results of these studies with evidence from epidemiological and clinical data, well-studied animal models and in vitro experiments utilizing specific formulations and variables, including the time of initiation of therapy, the length of treatment, the age of the population, etc. (reviewed in(4)). There exists now a great need for an intense and honest bidirectional communication between clinicians and basic science researchers to make sense of the divergent results of the WHI and WHIMS trials.
As a result of intensive research in the past 5-6 years reanalyzing the WHI trial data, as well as recent ancillary studies and many observational studies, a consensus has already been reached indicating that, among many factors, ET is safer than HT and that the “timing” could be a significant regulator of the health impact of HT on postmenopausal women. These data clearly show that unopposed ET, if taken within ten years after the onset of the menopause, did not increase the incidence of breast cancer but in fact provided some protection. On the contrary, ET initiated later (i.e., longer than ten years after the onset of the menopause) increased the risk of breast cancer.(5)(6)(7)
Recent data, although not unanimous, also indicate that ET, if initiated around the time of menopause, but not when started decades after the onset of the menopause, decreases cognitive decline in aging women (for a recent review see Sherwin(8)). Therefore, it seems that the ‘critical period or window” hypothesis can be applied for ET to prevent or delay cognitive decline in aging women. These observations also suggest that ET appears to have a “healthy cell bias” for neuronal action,(9) i.e., it has the potential as a preventive rather than as part of later treatment for memory loss, cognitive decline and neurodegenerative diseases such as Alzheimer’s (AD) in postmenopausal women.(10)
However, stroke, probably due to the elevated coagulation and inflammation caused by estrogen, remains a serious risk, regardless of the time from the onset of menopause or the form of HT (estrogen alone or in combination with progestins).(11)
Because the brain is one of the most important targets for estrogen and the side effects of ET occur primarily in the periphery (uterus, breast, blood and heart/vessels), this Cyberounds® will summarize efforts to introduce novel ETs to provide beneficial effects in the central nervous system (CNS) without exhibiting side effects in the periphery. Such ET would alleviate menopausal symptoms (hot flashes, cognitive and memory loss, sleep disturbances, depression and anxiety) and provide benefits in neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, traumatic brain and spinal cord injury and cerebrovascular stroke, without having any actions in the periphery.
These novel ligands are often referred to as CNS-selective estrogens but the designation as neuro-SERM, phyto-SERM or non-feminizing estrogens can also be found in the literature. In this Cyberounds® review, we will focus on three approaches to reach this goal: (i) develop novel ERβ-specific ligands and/or use phytoestrogens which have higher affinity for ERβ than ERα; (ii) develop non-feminizing estrogens and (iii) utilize prodrugs of estrogens from which the release of the biologically active hormone can be confined to the CNS. However, before discussing these approaches, we will provide a short update on estrogen action with specific emphasis on the CNS.
Mechanisms of Estrogen Neuroprotection
Estrogen provides neuroprotection by several mechanisms that impact neuronal survival: (i) regulation of transcriptional activity via the classical genomic action in the cell nucleus where the liganded ER binds to estrogen response element (ERE) of the promoter regions of targeted genes (Figure 1); (ii) regulation of transcriptional activity via the control of second messenger signaling cascades (kinases and phosphatases) initiated by membrane-associated ERs (Figure 1); (iii) regulation of cell survival by ER signaling initiated in the mitochondria; (iv) regulation of the physicochemical characteristics/conductivity of ion channels associated with neurotransmitter receptors; and (v) antioxidant activity via ER-independent mechanisms.
ER-dependent Classical Genomic Pathways
The genomic pathway involving the binding of estradiol to the cognate nuclear ER followed by gene transcription(12) has been implicated in many CNS actions including the promotion of neuronal survival. The ligated ERs can form homodimers (ERα/ERα or ERβ/ERβ) or heterodimers (ERα/ERβ) that can bind to the estrogen-response element (ERE) of the nuclear DNA. The dimers can interact with numerous coregulator proteins (co-activators and co-repressors).(13) These proteins participate in and recruit many enzymatic and structural proteins that allow modulation of chromatin structure to facilitate stimulation or repression of gene expression.

Figure 1. Classical (nuclear) and Rapid Intracellular Signaling By Estrogen Receptors (ERs).
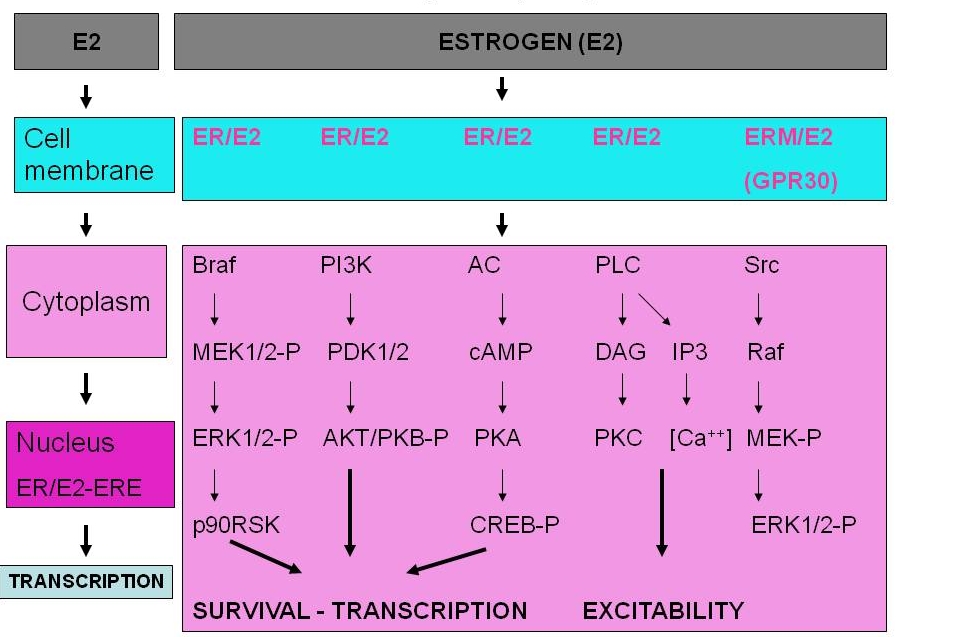
Click image for full size.
In the classical mode, estrogen penetrates the membrane and the cytoplasm and enters the nucleus where it binds to ER. The ligand-bearing ER (E2/ER) binds to estrogen response element (ERE) of target genes and regulates gene transcription. In the rapid mode of action, estrogen binds to membrane-associated ER or to a G protein-coupled receptor (e.g., GPR30) and activates intracellular signaling resulting in the promotion of cellular survival, the regulation of transcription and excitability.
Non-genomic Mechanisms of Action
In addition to the classical genomic pathways, estrogens elicit their CNS-action by a variety of non-genomic mechanisms.(14)(15)
ER-dependent Rapid Intracellular Signaling
Estradiol rapidly induces a variety of cellular responses that cannot be reconciled with the delayed genomic activity of the hormone. In addition to nuclear ERs, the existence of membrane/cytoplasmic pools of ERα and ERβ has recently been demonstrated (reviewed by(16)(17) and the ligand-bound ERs can function as a cytoplasmic signaling molecule involved in neuroprotection.(18) Specifically, ERα has been shown to bind in a ligand-dependent manner to the p85 alpha regulatory subunit of phosphatidylinositol 3-kinase (PI3K).(19) Therefore, stimulation with estradiol increases ERα-associated PI3K activity, leading to the activation of protein kinase B/Akt and endothelial nitric oxide synthase (eNOS).
The mitogen-activated protein kinase (MAPK) cascade(20)(21) and the cyclic-AMP-responsive element binding (CREB) protein signaling pathway(22) also respond rapidly to estradiol and have been implicated in its neuroprotective effects. Upstream mechanisms that initiate the signaling cascade leading to estrogen-inducible neuroprotection may be associated with rapid Ca2+-influx, which activates Src/extracellular signal-regulated kinase (ERK) and cAMP signaling pathways, and induces Bcl-2 protein expression (Figure 1).(23)
Estradiol can directly influence neurotransmission by binding to various transmembrane ion channels.(24)(25). Moreover, estradiol can provide neuroprotection by maintaining functionally intact mitochondria via mitochondrial ERβ.(26)(27) In addition, GPR30 (a G protein-coupled receptor uniquely localized to the endoplasmic reticulum) has been found to bind estradiol. Its activation results in intracellular Ca2+-mobilization and synthesis of phosphatidylinositol 3,4,5-triphosphate (IP3), the activation of the MEK-ERK(28) and, as mentioned above, activation of PI3K ultimately leading to neuroprotection.
However, modulation of intracellular pathways may occur not only through the binding of estradiol to ER(19) but also independently of ligand binding.(29)(30) For example, estradiol can also activate intracellular pathways independently of ERs or simply provide neuroprotection as a free-radical scavenger.
Antioxidant and Antiinflammatory Effects of Estrogen
Neuronal damage by free radicals has been implicated in most neurodegenerative diseases(31)(32) and aging.(33)(34) Estradiol's ER-independent antioxidant effects(35)(36) are mainly due to its direct free-radical scavenging ability,(37)(38) although indirect mechanisms, such as upregulation of antioxidant enzymes(39) and chelation of redox-active metal ions,(40) may also be involved.
In summary, neuroprotective effects of estradiol involve ER-mediated mechanisms (the regulation of genes that promote neuronal survival or promote cell death) and ER-independent (non-genomic) activity. ER-mediated mechanisms include the classical genomic action in the cell nucleus and mechanisms initiated by ER-signaling from the plasma membrane or mitochondria. Finally, the ER-independent mechanisms include the regulation of the activity of ion channels associated with neurotransmitter receptors and the antioxidant activity of estrogen.
Figure 2 summarizes these estradiol-regulated cell survival and death promoting mechanisms. As shown, estradiol stimulates the expression of many genes which are involved in the promotion of cell survival or neurogenesis including neurotrophic factors such as nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophins 3 and 4 and insulin-like growth factor 1 (IGF-1),(41)(42) as well as their receptors.(43) Additional target genes implicated in estradiol-mediated neuroprotection are associated with apoptosis. Apoptosis is the regulated form of cell death to remove unneeded, damaged or potentially deleterious cells(44) and plays a central role in development and homeostasis.(45) By the genomic mechanism associated with apoptosis, estradiol may rescue neurons through the induction of antiapoptotic proteins, such as Bcl-2(46)(47) and Bcl-x,(48) or down-regulation of apoptotic proteins, such as Bcl-2-associated X protein (BAX).(49) Estradiol may also induce several gene products that maintain cellular architecture such as neurofilament(50) and microtubulin-associated proteins.(51)
Contrary to supporting these survival-promoting mechanisms, estradiol also interferes or down-regulates the expression of genes involved in promoting cell death such as the induction of inhibitors against the death executing caspases.(52) Estradiol has also been shown to mediate inflammatory events. Specifically, it suppresses the induction of the cyclooxygenase-2 pathway mediated by interleukin-1β (IL-1β) in cerebral blood vessels and prevents migration of microglia into the CNS following inflammatory challenge.(53) Brain macrophages such as microglia cells have also been implicated in estradiol's action on the brain. Estradiol prevents the activation of microglia and the recruitment of peripheral monocytes induced by intraventricular injection of lipopolysacharide. This effect occurs through ERα activation and reduces the expression of neuroinflammatory mediators such as the inducible form of NO synthase (iNOS), prostaglandin-E2, the matrix metalloproteinase 9, lysosomal enzymes and complement C3 receptor, as well as by preventing morphological changes occurring in microglia during the inflammatory response.(53)(54) Estradiol also decreases microglial superoxide production and phagocytic activity by both an ER- and MAPK-dependent pathway(55) and it inhibits pro-inflammatory gene expression by controlling intracellular localization of nuclear factor-κ B (NF-κB(56)). Interested readers are referred to comprehensive reviews by(15)(37)(57)(58) for expanded coverage of non-genomic mechanisms of estrogen neuroprotection.
Figure 2. Summary of Survival- and Death-promoting Mechanisms Affected By Estrogen.
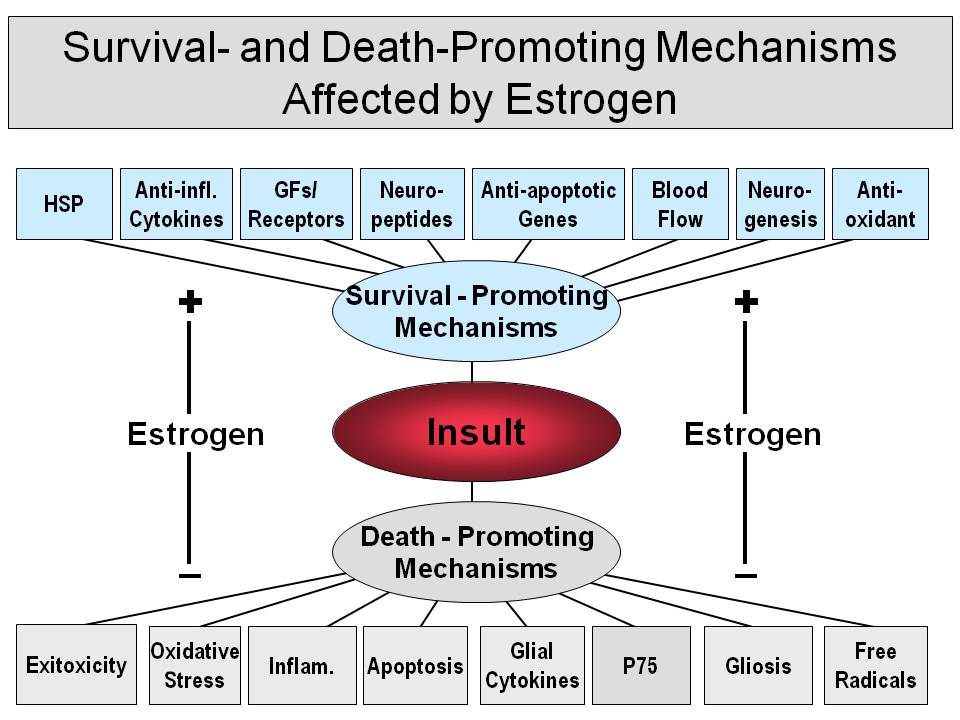
Click image for full size.
For details, see text.
Efforts to Develop CNS-selective Estrogen Therapy
ERβ-selective Ligands
Initial studies in rodents(59) provided great hope for developing ERβ-specific ligands for CNS-related diseases since ERβ was present in many brain areas involved in the pathogenesis of Alzheimer's disease, dementia, depression, etc., such as the cortex, hippocampus and the mesencephalic raphe nuclei, respectively, but was absent or barely expressed in the uterus, pituitary and the breast. Therefore, an ERβ-selective ligand that binds preferentially to ERβ over ERα could enhance neural responses without affecting the reproductive system. Follow up studies utilizing animal models provided further reinforcement for ERβ as a rational target for CNS diseases.
First, ERβ has been shown to play a crucial role in mediating estrogenic activities against neurodegeneration(60)(61) and promotion of neural synaptic plasticity, learning and memory.(62)(63) In addition to its cognitive role, researchers speculate that ERβ could be a novel therapeutic target for a range of pathologic conditions including brain disorders such as anxiety(64) and depressive behavior.(65) Second, in view of the possibility that simultaneous activation of both ERα and ERβ by their cognate agonists may diminish their overall efficacy,(60) an ERβ-selective agonist could reduce the antagonistic interactions among ERα and ERβ-selective agonists including the most efficacious endogenous 17β-estradiol. Third, selective activation of ERβ avoids ERα-mediated proliferative responses known to cause elevated risk for reproductive cancers in the uterus and breast associated with the use of the conventional HT in women.(66)
The distribution of ERβ, however, is somewhat different in primates from the pattern seen in rodents, as ERβ is expressed in the uterus and the breast, although, as in rodents, ERα is the major isotype in these organs.(67) Therefore, although continuing interest to develop ERβ-selective ligands for different CNS-related diseases exists, the initial enthusiasm is diminishing. However, the development of an ERβ-specific ligand with preferred CNS distribution (optimal or close to the optimal lipophilicity for blood-brain barrier penetration, etc.) is still desirable and feasible.
Plant-derived phytoestrogens, a weaker form than mammalian estrogens, produce estrogenic/antiestrogenic effects depending on the status of endogenous estrogens and the distribution of two subtypes of ERs in specific tissues.(68) It has been suggested that the low incidence of a number of sex hormone-related disorders, such as menopausal hot flashes and breast cancer in Asian women and prostate cancer in Asian men, compared to the incidence in Westerners, is partially attributable to the higher intake of phytoestrogen-rich soy foods in Asians.(69) The high intake of soy-derived phytoestrogens has also been linked to the low prevalence rate of AD in Asia compared to the Western world. This association, however, lacks confirmation from randomized and controlled human studies, which have been found to be inconsistent.(68) More importantly, these studies evaluated the efficacy of many soy products but not of pure S-equol.
Equol is an isoflavone that is formed by the biotransformation of daidzein by intestinal microflora in the gut of certain individuals, particularly Asians(70)(71)(72) (Figure 3). It is a chiral (lacking an internal plane of symmetry) molecule existing as R- and S-enantiomer(73), the latter having a 8-10 fold selectivity for estrogen receptor-beta (ERβ)
Figure 3. Biotransformation of Diadzein to S-equol.
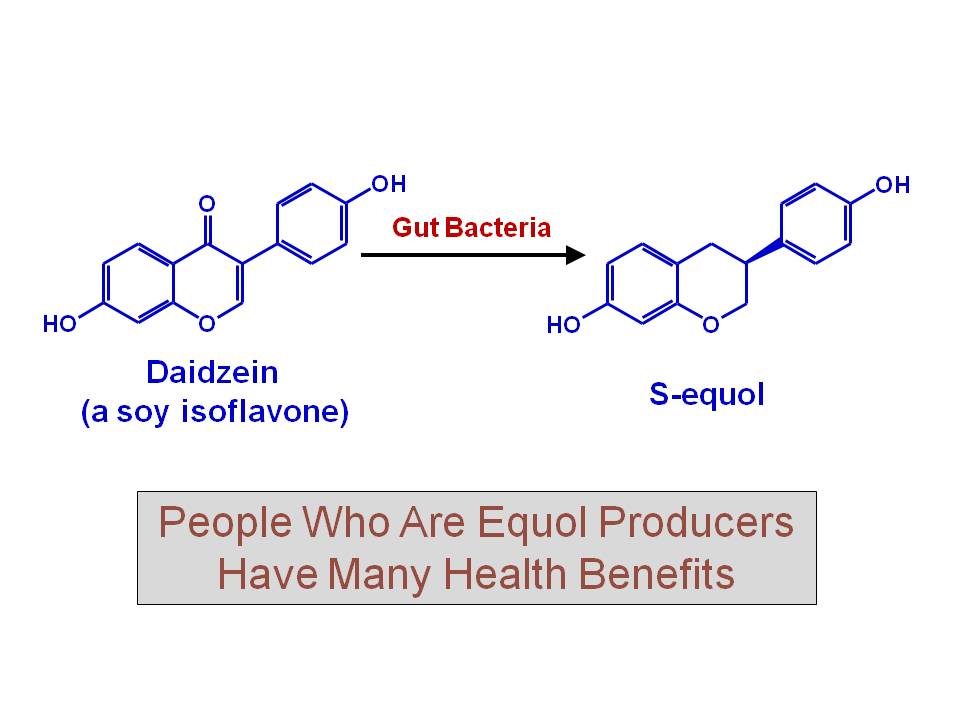
Click image for full size.
Recently, Ishiwata et al.(81) reported that non-equol producer Japanese women given a nutraceutical containing S-equol showed improved mood-related symptoms (decrease in depression, anxiety and fatigue, vasomotor symptoms and improvement in vigor) without affecting LH and FSH levels. These observations are interesting as they suggest that S-equol acts as an ERβ-selective ligand in humans because mood regulation by estrogen is believed to be mediated via ERβ and not via ERα. In animal models, estrogen can exert both anxiolytic and anxiogenic actions depending on the behavioral context. However, ERα-selective ligands such as 1,3,5-tris(4-hydroxyphenyl)-4-propyl-1H-pyrazole (PPT) are anxiogenic, while ERβ-selective compounds such as 2,3-bis(4-hydroxyphenyl)propionitrile (DPN) are anxiolytic and anti-depressive.(64)(82)(83) These human and rodent observations, together with the well known phenotype of the ERβ null mice, i.e., mice characterized by increased anxiety in conjunction with lower serotonin content in the dorsal raphe nucleus, suggest that estrogen improves mood by acting via ERβ in humans, and S-equol might exert beneficial effects not only in hot flashes but on anxiety/depression as well.
Our recent findings with the rat model show that S-equol alleviates hot flashes (tails skin temperature [TST] rise) and provides bone protection in ovariectomized rats without stimulating the uterus. These observations are surprising as previous studies failed to show efficacy of ERβ-selective ligands in this model,(84) while the ERα-selective compound (PPT)(85) did blunt hot flushes (TST rise in rats),(86) suggesting that hot flushes could be prevented by ERα- but not by ERβ-selective ligands. The observation by Ishiwata et al.(81) that S-equol, an ERβ-selective ligand, also prevented hot flashes in equol-producer menopausal women suggests that both ERs are involved in thermoregulation, specifically in the pathomechanism of hot flashes.
Other rodent studies also concluded that racemic (S/R-) equol stimulates uterine wet weight only at high, pharmacological doses but not at low, physiological doses. At high dose, equol also presented histological features of mild estrogenic stimulation and increased the expression of estrogen-regulated genes in the uterus including progesterone receptor (PR), proliferating cell nuclear antigen (PCNA), complement-3 (C3) and insulin-like growth factor 1 (IGF-1).(87)
Estrogen Derivatives and Analogs With Reduced Or No Affinity To ER
The approaches to develop neuroprotective estradiol analogues or derivatives that reduce or eliminate genomic effects, but retain the non-genomic effects, such as the anti-oxidant effects of estradiol, include pharmacological manipulations of estrogens resulting in “estrogen-like” compounds which do not function anymore as the parent hormones. These analogues and derivatives, often referred incorrectly to as non-feminizing estrogens,(88) are expected to curtail endocrine side effects associated with chronic estrogen therapy(2)(89) that impacts ER in the liver, uterus, breast, and other tissues. In addition, they would also allow for treatment of both women and men.
Estrogens, including the natural 17β-estradiol, estrone, estriol, etc., are highly lipophilic molecules and concentrate in lipid-rich regions of the cell, such as cellular membranes and myelin(27) affecting membrane fluidity.(90) In addition to binding to ER in the cellular membrane, cytosol and nuclear compartments, they may also interact with lipid-transport proteins.(91) Therefore, it is likely that estrogens act in vivo as highly localized antioxidants.(38)
The finding that the ER-α/β ligand 17β-E2 and its enantiomer 17α-E2, which binds to these ER isoforms with 40-fold lower affinity, were equally potent in protecting neuronal cells, while the effects were not blocked by an estrogen antagonist,(92) prompted a closer look at the structural requirements for neuroprotection among steroids.(93)(94) All estrogens tested with an intact phenolic A ring were neuroprotective including 17β-E2 and 17α-E2, as well as estrone (E1), estriol (E3), the potent synthetic estrogen ethinylestradiol (EE) and the catechol estrogens(95) representing the principal cytochrome P450 metabolites of estrogens. However, 3-O-methyl(94) and 3-O-butyl ether congeners(96), 3-O-cycloalkyl ether, and 3-O-acetyl derivatives were found inactive, indicating the importance of a phenolic A ring. Steroids that lack a phenolic A ring (testosterone, dihydrotestosterone, progesterone, corticosterone, prednisolone, 6α-methylprednisolone, aldosterone and cholesterol), phenol, lipophilic phenols and tetrahydronapthol also were inactive.(94) Therefore, a phenolic A ring and at least three rings of the steroid nucleus were found necessary for the neuroprotective activity. This requirement was suggested as a basis for the design and synthesis of estrogen derivatives in which the structural modification would render the compound non-estrogenic.(93)
In the most comprehensive structure–activity study to date, estratrienes (over 70 compounds) were tested in vitro for neuroprotection and determined EC50 values to ascertain potency comparisons with 17β-E2. A few of these derivatives, which did not bind to either ERα or ERβ, have also been evaluated in vivo using ischemia-reperfusion injury induced by temporary middle cerebral artery occlusion in rats.(88)(97) Infarct volume was significantly reduced, whereas cerebral blood flow was increased compared to ovariectomized control animals. The magnitude of these beneficial effects was similar to that of the endogenous estrogens. Altogether, these compounds possessed both neuroprotective and vasoactive effects, which offered the attractive possibility of clinical application for stroke or other neurodegenerative diseases such as AD, PD and aging, where free-radical generation and oxidative stress are part of the pathomechanism without the side effects of estrogen. However, the most potent derivatives were larger in molecular weight/size and significantly more lipophilic than 17β-E2(97); hence, they were not suitable for potential drug development.
Brain-targeting Prodrugs of Estrogens
Bioreversible modification by prodrug design of the naturally occurring estrogens to target the action of the steroid into the CNS and reduce or even eliminate systemic side effects has been considered. This alternative strategy is not burdened by the excessive costs, enormous challenges and high risks associated with an extensive screening and discovery program to find “neuro-SERMs” with an optimal activity profile to replace estrogens as therapeutic entities. Prodrugs are inactive precursors of the therapeutic agents that are converted to the biologically active agents by enzymatic and/or chemical transformation in vivo – preferably at the site of action.(98)(99) The application of prodrugs (which often have improved physicochemical properties compared to the parent drug) to overcome barriers to a drug's usefulness and improve the therapeutic index is an established method.(100)(101)
The concept of targeting estrogens to the brain by conjugating them with the dihydrotrigonelline-trigonelline redox carrier was considered a promising prodrug approach (also referred to as “chemical delivery system,” CDS) that advanced to the clinic.(102) The 1,4-dihydrotrigonellyl 17β-O-ester of the female hormone has been considered, among others, for the potential treatment of Alzheimer's disease and stroke(103) a potential alternative to traditional ET.(104) As summarized schematically in Figure 4, CDSs for estrogens are essentially “pro-prodrugs” that require two metabolic steps to produce the parent hormone.(105)The attachment of 1,4-dihydrotrigonellyl as a pro-moiety provides an increase in lipophilicity favoring penetration across the blood-brain barrier (BBB), and also is believed to promote retention in the CNS. This is due to an in situ metabolic conversion of the non-ionic dihydropyridine to a membrane-impermeable (positively charged) pyridinium ion as an intermediate before the release of E2.
The oxidative conversion of a dihydropiridine to pyridinium occurs ubiquitously, and it is analogous to the oxidation of NAD(P)H, a coenzyme associated with many oxidoreductases and cellular respiration. Any of the oxidized form in the periphery is presumed to be rapidly lost, as it is polar and thus an excellent candidate for elimination by the kidney and bile. Concentration of the active drug is presumed to remain low in the periphery and, thus, expected to reduce systemic, dose-related toxicities. Essentially, the CDS strategy was designed to function as a targeting system because a preferential delivery of the active agent to the CNS versus the rest of the body might be achieved. Finally, the CNS-retained compound is expected to release the active compound through subsequent metabolic processes (usually enzymatic hydrolysis) in a sustained manner. Several practical hurdles, however, have hindered pharmaceutical development and acceptance of CDS: cumbersome chemistry affecting the drug’s production and purity,(106) formulation problems because of its insolubility in water and limited stability,(107) uterotrophic side-effects(108) and the potential toxicity of its pyridinium carrier moiety(109) designed to be released together with the estrogen in the brain.
Figure 4. Schematic Illustration of Brain-targeting By Chemical Delivery System for Estradiol.(105)
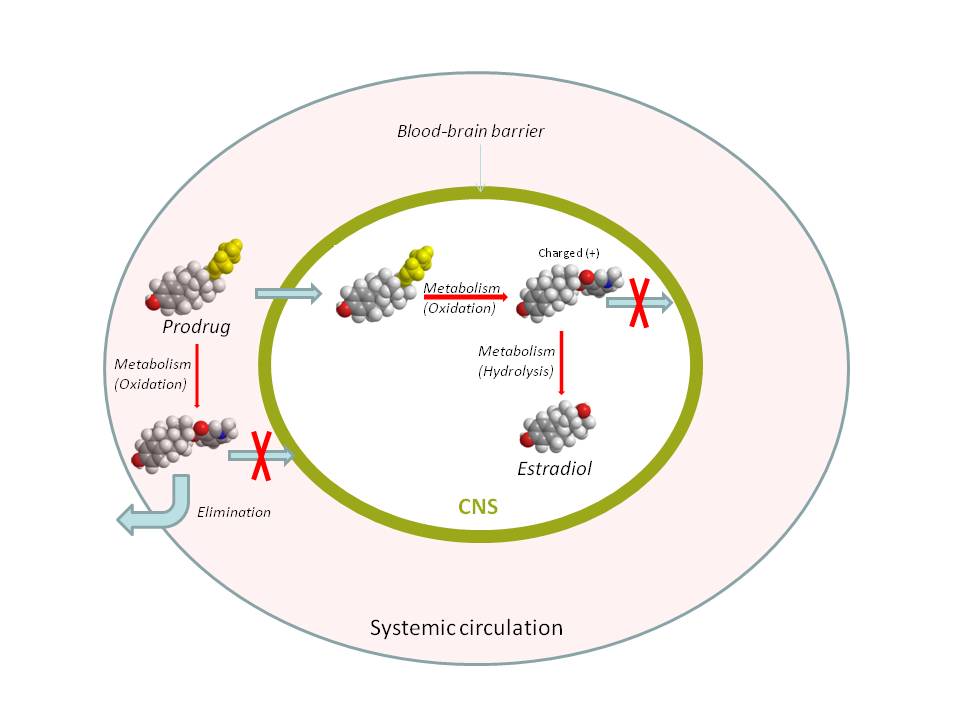
Click image for full size.
Another approach to CNS-targeting of the hormone may rely upon, along with optimizing physicochemical properties to cross the BBB,(110) a preferential bioactivation of an estrogen prodrug in the target tissue versus the rest of the body. As shown schematically in Figure 5, a preferential conversion of the prodrug to the hormone would create a powerful metabolic “sink” and, thus, concentrate the formation of estrogen to the brain. Estrogen-derived para-quinols have been recently identified as prodrugs (“bioprecursors”) that confine the hormone’s action primarily to the brain by this mechanism,(111) while their conversion to estrogens has been shown to produce no toxic by-products and no induction of oxidative stress.(112)(113) Proof-of-concept studies for maintaining CNS-effects upon treatment with an estrogen-derived para-quinol prodrug without systemic effects (manifested by the lack of uterine stimulation) have been provided in a stroke model employing focal ischemia(111) and the rat hot flash model.
Figure 5. Concept of Brain-selective Estrogen Prodrug Based on Preferential Metabolism. (111)
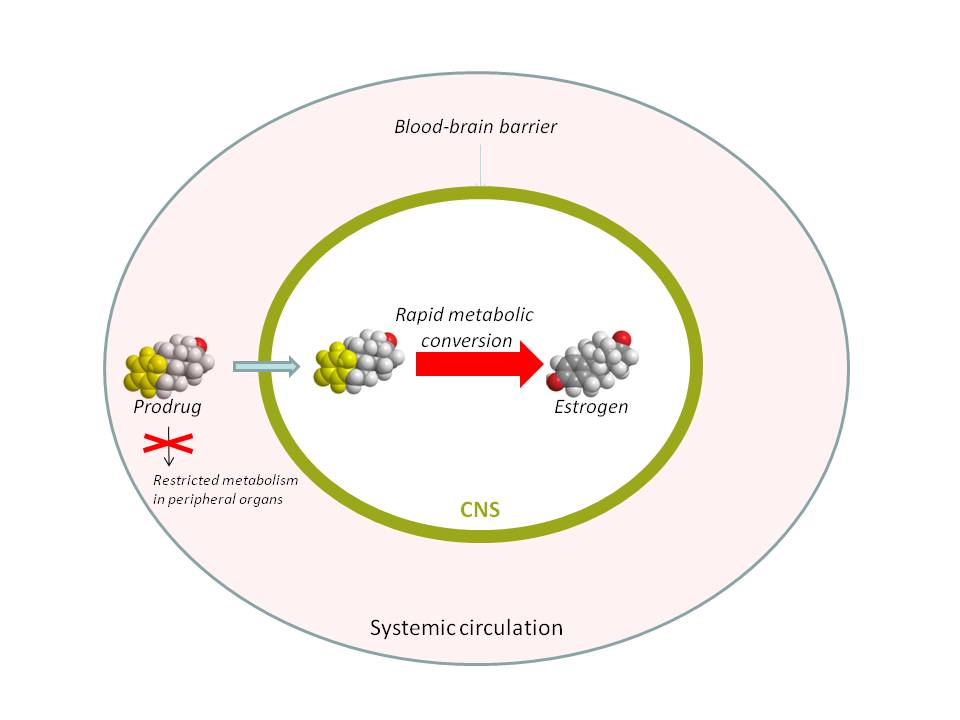
Click image for full size.
Conclusions
Although the WHI/WHIMS trials raised serious concerns about ET, reanalysis of the data from these studies indicated that some of the concerns can be prevented if the right research study decision is made on the population included, drugs of choice, doses and route of administration, age at which hormone exposure occurs or timing of ET or HT initiation in relation to the onset of the menopause “critical period” or “window”.
At the same time, the WHI and WHIMS studies also provided the impetus to the scientific community to challenge and reconcile the results of these studies and develop novel CNS-selective estrogens that did not provoke side effects in the periphery, i.e., they would not have proliferative effects in the breast and uterus, and would have no effect on coagulation. Among the three approaches (ERβ-selective ligands or phytoestrogens, neuro-SERMs and pro-estrogens) reviewed, the prodrug approach seems to be the most likely to succeed, promising, in our opinion, to give the full benefits expected from CNS-selective ET/HT.
Acknowledgments
The authors’ research in the subjects covered was supported in part by grants (AG031535 to I.M. and L.P., NS044765 to L.P.) from the National Institutes of Health and by a grant to I.M. from Ausio Pharmaceuticals. L.P. acknowledges endowment (BK-0031) from the Welch Foundation.