Course Authors
Robert J. Pignolo, M.D., Ph.D.
Dr. Pignolo is Assistant Professor and Director, Ralston-Penn Clinic for Osteoporosis & Related Bone Disorders, Department of Medicine, Division of Geriatric Medicine, University of Pennsylvania School of Medicine, Philadelphia, PA.
Within the past 12 months, Dr. Pignolo reports no commercial conflicts of interest.
Albert Einstein College of Medicine, CCME staff and interMDnet staff have nothing to disclose.
Estimated course time: 1 hour(s).

Albert Einstein College of Medicine – Montefiore Medical Center designates this enduring material activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
In support of improving patient care, this activity has been planned and implemented by Albert Einstein College of Medicine-Montefiore Medical Center and InterMDnet. Albert Einstein College of Medicine – Montefiore Medical Center is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Upon completion of this Cyberounds®, you should be able to:
Define characteristics of cellular aging
List the key mechanisms responsible for cell senescence
Discuss the relationship between cell aging in vitro and in vivo
Describe the relationship between cell senescence and age-related diseases.
The term senescence is often used to describe aging at the cellular level, and after Hayflick’s original observations on the limited in vitro life span of normal human cells in culture,(1)(2) aging cells have often been referred to as undergoing replicative senescence. The life span of cells are usually measured in passages or, more accurately, in population doublings. The “age” of cells can be determined retrospectively as the percentage of life span completed from the point at which there is negligible cell division (e.g., cells that have undergone 25 doublings, and ultimately undergo 50 doublings, can be operationally described as 50% life span completed). Alternatively, the age of cells can be determined prospectively as the percentage of cells able to undergo DNA synthesis during a period of time equivalent to the longest cell cycle time, usually 24−30 hours.(3) Descriptive terms such as “early” or “late” passage or population doubling, or “young” and “old” are often employed to designate the relative age of cells in culture.
The Phenotype of Aging Cells
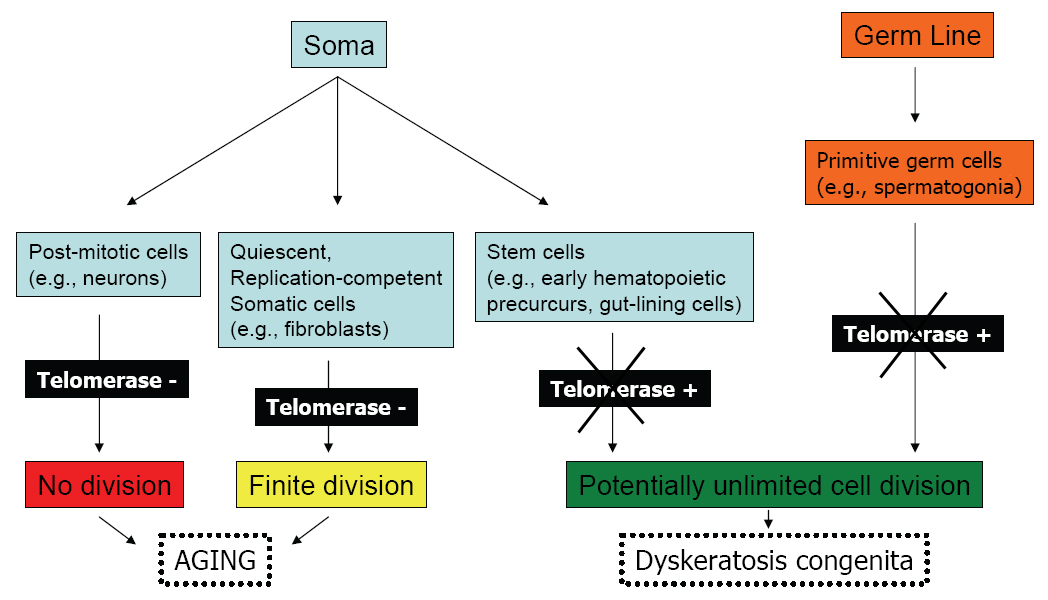
The definition of cell aging based on finite proliferative capacity is limited to those somatic cells that normally maintain their ability to divide in the organism and does not include germ line or certain stem cells which maintain an indefinite life span (Figure 1). Although replicative senescence has also been applied to animal cells in culture, particularly rodent−derived cells, in this situation there are the confounding variables such as "crisis" (or the mass loss of cells in culture) and spontaneous immortalization.

Figure 1. Replication Potential of Normal Human Cells.
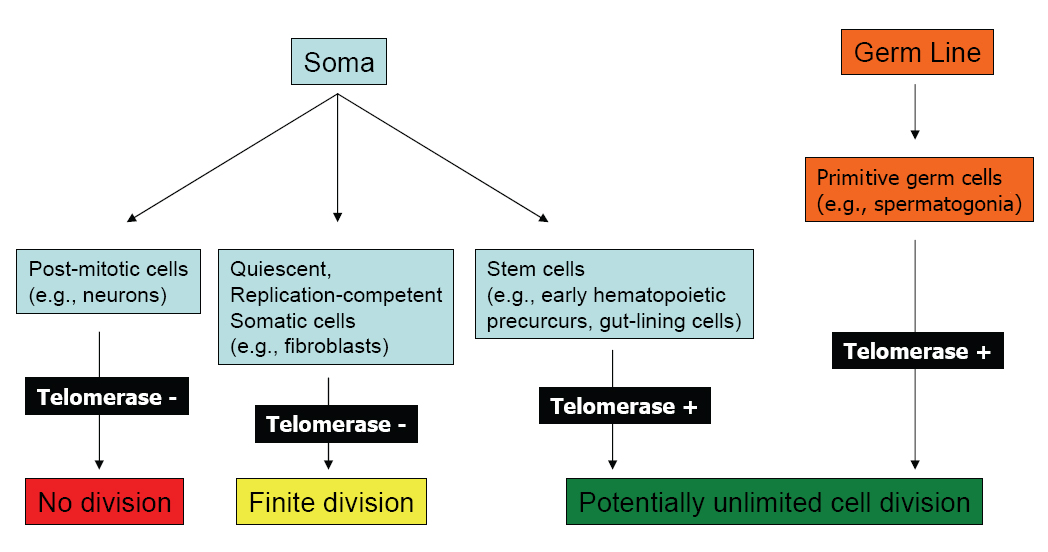
Click above image to view full size.
Shown are cell types derived from the soma or germ line, their telomerase status, and replicative capacity.
Characteristics of an aging cell (or population of cells) in culture are mostly assumed as a function of replications, not time in culture. Cells can be kept in culture for an indefinite period of time in a quiescent (non−dividing), but metabolically active state, and only when they are induced to divide do the changes that we describe as replicative cell senescence occur. Besides an essentially irreversible growth−arrested state, other characteristics of senescence include apoptosis resistance (especially in fibroblasts),(4)(5)(6) altered gene expression (both of proliferation−related genes as well as of those unrelated to growth arrest),(7)(8)(9) as well as biomarkers that in the appropriate context may identify replicatively−aged, damaged or stressed cells.(10)(11)(12)(13)(14)(15)
Caveats and Limitations
Most of the studies on which knowledge of mechanisms of cell senescence is based have been performed on fibroblasts or fibroblast−like cells, and thus may not be generally applicable to other cells types. Studies in mammals are also limited, but there is growing evidence that cell senescence does occur in vivo with aging in general, as well as in specific examples of age−related pathology. However, cell senescence is not always replicative, since post−mitotic cells, such as neurons, do age. Finally, the aging of stem cells with long or indefinite life spans, as well as aging of cells outside the mesenchymal lineages (such as that of hematopoietic cells and their precursors), may also be different from that described for fibroblasts.
Arrest State of Senescent Cells
As cells age in culture as a function of the number of divisions, they undergo several discrete stages. There is an initial stage of outgrowth from explant, a stage of vigorous proliferation, a plateau phase of growth, a loss of cell number characterized by non−apoptotic cell death, and the survival of a stable but non−dividing population of cells (Figure 2). Spontaneous transformation may occur, though it is an extremely rare event in human cells.
Figure 2. In Vitro Stages in the Life History of Normal Human Diploid Fibroblasts.
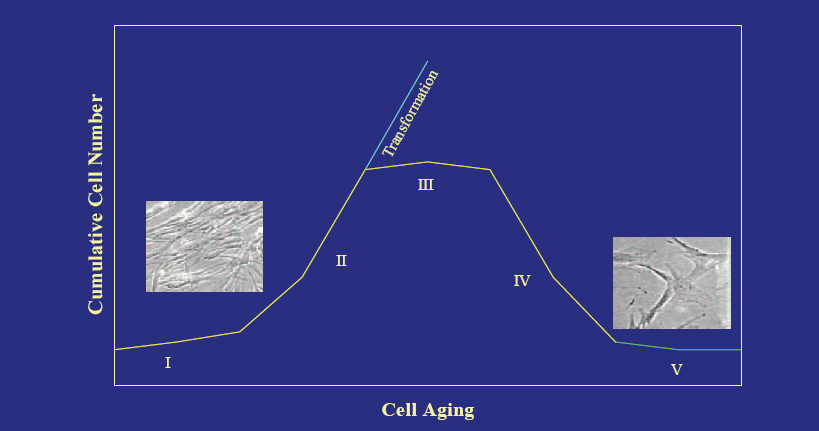
Click above image to view full size.
The initial stage of outgrowth from explant (I) is followed by a stage of vigorous proliferation (II), a plateau phase of growth (III), a loss of cell number characterized by non-apoptotic cell death (IV), and the survival of a stable but non-dividing population of cells (V).
As cells become proliferatively senescent, cell cycle times progressively increase. This occurs largely at the expense of a longer and longer G1 interval until, finally, senescent cells stop dividing with characteristics of a late G1 arrest (Figure 3).
Figure 3. Arrest State of Proliferatively Senescent Cells.
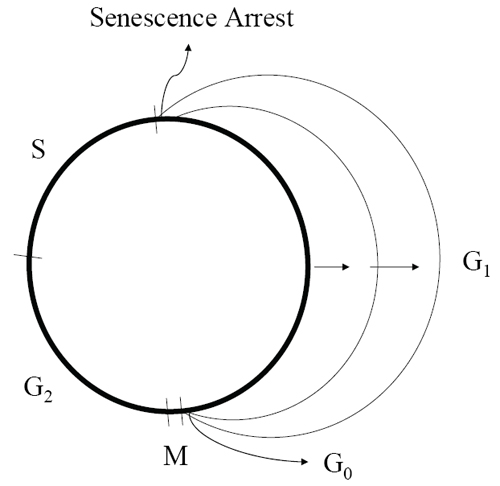
As cells undergo exhaustive replications, cell cycle times progressively increase, largely at the expense of a longer and longer G1 interval until, finally, cells stop dividing with characteristics of a late G1 arrest. G, Growth stages; S, DNA Synthesis; M, Mitosis
The evidence of a late G1 arrest is based on the ability of senescent cells to complete some of the mitogen−induced late G1 events as well as changes in chromatin condensation patterns typical of late G1.(16) Senescent cells are not quiescent, and are unable to obtain a true G0 growth−arrest state, as proliferatively competent cells do with withdrawal of growth factors or contact inhibition.(17)(18)(19)
Mechanisms of Senescence
Transcriptional regulators p53 and the retinoblastoma protein pRB mediate pathways that can induce and maintain the senescent state. These factors govern two cell−cycle cyclin−dependent kinase inhibitors (CDKIs), p21 (also named CDKN1a, p21Cip1, Waf1, or DS11) and p16 (alternatively named CDKN2a or p16INK4a).(20)(21)(22)(23). Although it is clear that p53 can directly upregulate p21, it is less well understood how p16 is induced. Both p21 and p16 keep pRB in an active, hypophosphorylated state but, depending on the insult that causes their induction and on the cell type in which they are induced, they may have independent or complementary effects (Figure 4).
Figure 4. Senescence Response and Escape From Senescence.
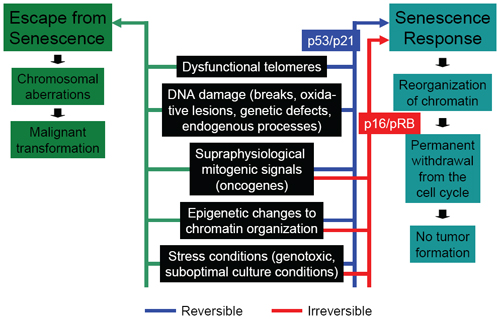
Comparison of p53/p21 and p16/pRB pathways in response to various promoters of senescence and their relationship to neoplastic transformation is shown.
Although somewhat of a generalization based on data in multiple cell types, the p53/p21 pathway activates a robust senescence response as the result of dysfunctional telomeres, DNA damage, supraphysiological mitogenic signals and oncogenes, epigenetic changes to chromatin organization, and stress conditions.(12)(14)(24)(25)(26) The p16/pRB pathway can be involved after many of the same cellular insults; however, in the case of dysfunctional telomeres and DNA damage, its specificity may be less and its kinetics may be delayed.(27)(28) Another difference between the p53/p21 and p16/pRB pathways may relate to the reversibility of the ensuing growth−arrested state, with p16 induction more likely to be associated with irreversible arrest states (at least in culture).(29)
Telomere Dysfunction and DNA Damage
Telomere−dependent senescence occurs as the result of loss of repetitive DNA at the ends of chromosomes (Figure 5) due to the limitation of DNA polymerases to completely replicate these ends during each S phase.
Figure 5. Repetitive DNA Sequences (6-base-pair repeats) At the Ends of Chromosomes Compose Telomeres.

Mammalian telomeres are protected from degradation or fusion by a complex t-loop structure (not shown), but they may become uncapped and eventually critically short and dysfunctional with senescence.
Mammalian telomeres are protected from degradation or fusion by a complex t−loop structure,(30) but they may become uncapped and eventually critically short and dysfunctional. Dysfunctional telomeres precipitate a DNA−damage response that causes cell cycle arrest and, depending on whether damage can be repaired, may cause a reversible arrest, or an irreversible arrest (senescence).(12)(14)(15)(31)(32) Many proteins participate in the DNA damage response, some of which can be found at the site of DNA damage (particularly double−stranded breaks). Telomere dysfunction−induced foci (TIFs) are sites of DNA−damage along the telomere where certain proteins, including adaptor proteins and chromatin modifiers such as 53BP1 and γ−H2AX, respectively, are localized. Few dysfunctional telomeres per cell are required to trigger senescence.(33)(34)
Stress
Cells can exhibit a senescence response under conditions of stress, including genotoxic stress (e.g., free radical damage), supraphysiological mitogenic signaling, suboptimal or inadequate growth conditions, and sustained exposure to certain cytokines. Independent of telomere shortening, certain cells types can undergo a p16−mediated senescence in culture, presumably from the use of standard albeit inadequate growth media and conditions.(35) Oxidative stress, defined as the response to an oxygen tension above the level a cell in its usual (in vivo) environment normally experiences, can also induce p16.(36)(37) Increased intracellular oxygen radicals secondary to chronic stimulation by interferon−β, however, appear to promote senescence through a p−53 dependent DNA damage response.(38)
Oncogenes
Oncogenes, aberrant forms of genes normally involved in stimulation of proliferation and that increase the likelihood of cell transformation, can induce senescence in normal cells. For example, proteins in the RAS signaling pathway cause a senescent phenotype when expressed in their oncogenic forms or when simply overexpressed.(39)(40)(41)(42)(43) Oncogene−induced senescence occurs in the absence of telomere shortening but nevertheless causes a DNA damage response as a result of faulty DNA replication.(25)(44) The induction of growth arrest due to oncogenic signaling may also involve changes in chromatin structure characteristic of the senescent state.(13)(45)(46)
Chromatin Changes
Irreversible proliferative arrest can be associated with the reorganization of chromatin into discrete domains named senescence−associated heterochromatin foci (SAHFs), where inaccessibility to large stretches of DNA prevents transcriptional activation of genes. The mechanism of inactive chromatin formation is likely to be mediated by pRB, at least in some cell types that undergo senescence.(13)(47) It is thought that SAHFs inactivate genes that are important for cell−cycle progression, particularly those induced by the transcription factor E2F (e.g., cyclin A2). So−called histone chaperones, such as HIRA, ASF1a and UBN1, function as a complex to mediate chromosome condensation into SAHFs.(46)(48) This complex preferentially deposits the variant histone H3.3 over the usual histone H3.(1)(49)(50) although the important events in SAHF formation are still incompletely understood. It is paradoxical that inhibition of histone deacetylase by chemical methods, which produces an open chromatin conformation, also causes senescence.(51)(52)
Senescence and Immortalization
Normal human cells capable of finite replication lack the enzyme telomerase which functions to maintain telomere length. As described above, when telomeres grow critically short, cells undergo telomere−dependent senescence. This stage of growth arrest is sometimes referred to as the mortality 1 (M1) stage of senescence, essentially the first barrier to unlimited cell proliferation.(53)(54) In the presence of factors that can overcome this barrier by inactivating p53− or pRB−based pathways (e.g., certain viral antigens), cells can undergo additional rounds of replication but at the expense of even shorter telomeres. Subsequent to this additional proliferation, cells undergo a crisis state or M2 stage that results in the elimination of all but a few rare surviving cells.(53) These surviving cells acquire changes that result in their immortalization, often but not exclusively as the result of new telomerase expression.(55)
Biomarkers of Cell Senescence
Knowledge of potential mechanisms for senescence, altered gene expression with cellular aging, as well as serendipitous findings, have led to the use of various markers for identification of senescent cells, both in vitro and in vivo.
Dysfunctional telomeres may be related to cell and organismal aging based on several observations. Telomeres shorten with age in many human tissues. Telomere shortening or dysfunction can cause cell senescence in cultured cells. Particularly short telomeres and putative senescent cells (based on other criteria) are associated with sites of pathology (e.g., atherosclerosis, cirrhosis).(56)(57)(58)(59)(60) Individuals with short telomeres also have higher mortality rates.(61) Several human progeroid diseases have telomere defects and demonstrate premature cell senescence of cultured cells (e.g., Werner syndrome, ataxia telangiectasia, HGPS, dyskeratosis congenita).(62)(63) Uncapped telomeres accumulate to high levels (~15% of cells) in aged baboon skin.(64)
There are several arguments against a role for telomere dysfunction as a marker for aging. For example, telomere shortening cannot explain aging in post−mitotic tissues (e.g., neurons). Mice lacking telomerase do not age more rapidly, although they eventually develop disease.(65)(66) Humans with partial loss of telomerase (Dyskeratosis congenita) develop diseases that are not a simple picture of rapid aging.(67) Given these observations, it is likely that telomere dysfunction is most relevant to aging in those replication−competent somatic cells (e.g., fibroblasts) that always lack telomerase activity (Figure 6).
Figure 6. Telomere Dysfunction In Aging and In A Progeroid Syndrome Caused By Telomerase Deficit.
Click above image to view full size.
Normal aging phenotypes are likely due to changes in post-mitotic cells over time and in replication-competent somatic cells (e.g., fibroblasts) that always lack telomerase activity. Humans with partial loss of telomerase (Dyskeratosis congenita) develop conditions that result from stem cell or germ line defects and are not a simple picture of rapid aging.
Senescence−associated β−galactosidase (SA−β−gal) activity is considered a biomarker of replicative senescence; however, it does not easily distinguish senescent cells from early passage quiescent, or stressed cells.(68) SA−β−gal activity is reflected as a preferential hydrolase substrate cleavage at pH 6.0; it is not a unique enzyme but the lysosomal enzyme measured at suboptimal pH. SA−β−gal activity increases with replicative age, as well as in quiescent cells, serum−starved cells and cells in response to hydrogen peroxide treatment. Mixed findings have been reported in skin tissue from various aged donors.
EPC−1/PEDF (early population doubling level cDNA−1/retinal pigmented epithelium−derived factor) is a quiescence−specific gene that is a potent inhibitor of angiogenesis.(19)(69)(70)Its expression declines during replicative aging of cells in culture and in situ in skin sections from donors with increasing age.(71) p16 has been used to identify some senescent cells;(29)(72)(73) however, it is also expressed in some transformed cells.(74)
Senescence−associated heterochromatic foci (SAHF) are dense foci of heterochromatin that form in senescent cells and can be detected by preferential binding of DNA dyes such as 4’, 6−diamidino−2−phenylindole (DAPI). In human cells approaching senescence, the histone chaperone HIRA is transported into discrete nuclear foci called PML (acute ProMyelocytic Leukemia) nuclear bodies.(46) PML bodies are enriched in the protein PML and other nuclear regulatory proteins.(75)(76) In human cells, localization of HIRA to PML bodies is essential for formation of SAHF(48) and thus their detection could serve as independent biomarkers for senescence even prior to their translocation to chromatin and formation of SAHF.
Cellular Aging In Vivo
The notion that replicative senescence in vitro is a manifestation of aging at the cellular level is based on several in vivo correlative findings, not all of which have stood up to vigorous scrutiny. Cells from patients with segmental progeroid syndromes, so−called premature aging conditions that recapitulate some aspects or “segments” of physiologic aging, have a reduced in vitro proliferative capacity.(62)(63) Similarly, the clone size distribution of cultures derived from old individuals give rise to the lowest percentage of large colonies.(77)
A direct relationship between replicative capacity and species−specific maximum life span, as originally put forth by Rohme,(78) did not control for developmental status of donor cultures and contained inaccurate longevity data. A well−conceived follow−up study by Lorenzini et al. showed little correlation between fibroblast proliferative capacity and species maximum life span, and produced the interesting finding that proliferative life span is better correlated with body mass, especially among larger mammals.(79)
An inverse relationship between replicative life span and donor age had been demonstrated by several studies, but when pre−existing medical conditions are taken into account, no clear relationship was found to exist between replicative capacity and donor age, at least not for fibroblasts in mass culture.(80) The implications of these studies for replicative senescence and human aging are that (1) the health state of the donor influences replicative capacity; (2) proliferative ages of cells in vivo will likely vary depending on their replicative history such that two nearby cells may have different life spans based on the number of their previous divisions; and (3) aging in vivo may occur in a mosaic pattern rather than uniformly, whereby pathology in the organism may occur locally as a result of replicative senescence in only a few cells. In vitro studies of donor−derived mass cultures also tend to select for the fastest growing cells, and thus the extent of replicative senescence in vivo is likely to be grossly underestimated with the result of minimizing differences due to age alone.
Role of Cell Senescence in Age−related Pathology
Based on the ability to identify senescent cells in culture and in organisms, there is growing evidence that suggests functional relationships between cell senescence and tissue aging. There also appears to be important roles for senescent cells in age−related pathology, sometimes independent of limited replicative capacity.
The most well−established example of the involvement of senescence in age−related pathology is the relationship between cell aging and cancer. Evidence that telomere shortening/dysfunction inhibits carcinogenesis is suggested by the fact that insufficient telomerase leads to senescence and crisis, both barriers to cell division. The p53 and RB pathways, which enforce telomere− and oncogene−induced senescence, are typically mutated in cancers.(81)(82)(83)(84)(85) Also, cancer cells generally express high levels of telomerase. Further, high levels of telomerase immortalize cells, which otherwise do not immortalize spontaneously (or at least at a rate <~1/1010).
In a variety of cell types that secrete EPC−1/PEDF, including fibroblasts and retinal pigmented epithelial cells, there is a loss of expression with in vitro replicative senescence and with increasing age in vivo.(19)(71) In the eye, EPC−1/PEDF functions as an inhibitor of endothelial cell proliferation in the prevention of ischemia−induced retinopathy.(86) In a well−accepted murine model of ischemia−induced retinopathy, EPC−1/PEDF prevented tufts of endothelial cells breaking through the internal limiting membrane into the vitreous.(70) This has enormous implications for age−related eye disease such as diabetic retinopathy and macular degeneration and illustrates that there are changes in the secretory profile of senescent cells that contribute to age−related pathology. Other examples suggest that proteins secreted by senescent cells may stimulate the proliferation and angiogenic activity of local premalignant cells,(87)(88)(89)(90)(91)perhaps contributing to neoplastic progression of early cancers.
Cell senescence may also play a role in age−related bone loss. Reconstituting telomerase activity prevents telomere shortening and replicative senescence in human osteoblasts in vitro,(92) and telomerase expression in human mesenchymal stem cells actually enhances bone formation in vivo.(93)(94) Telomerase accelerates osteogenesis in these stem cells by upregulation of critical transcription factors such as Runx2 and osterix.(95) “Telomerized” presenescent osteoblasts can prevent bone mass loss in an in vivo transplant model.(96) In mouse models of premature aging based on telomere defects, osteoporosis is a major phenotype related to impaired osteoblast differentiation of mesenchymal precursor cells.(97)
In response to vascular injury, increased cell turnover is thought to be a key stage in the formation of atherosclerotic plaques. Since telomere shortening has an obvious association with cell turnover, Ogami et al. measured the telomere length of human coronary endothelial cells to test if there is a relationship between telomere length and coronary artery disease (CAD).(59) They found that shortened telomere lengths were present in endothelial cells from patients with CAD, suggesting that focal replicative senescence of endothelial cells may play a critical role in coronary atherogenesis.
Perturbation of the adaptive immune system with age results in a variety of pathophysiological manifestations. Diminished immune responsiveness, including decreased response to new antigens, decreased vaccine efficiency (e.g., influenza), and possibly compromised immune surveillance may become prominent with increasing age. Altered immune system physiology is illustrated by thymic involution, decreased production of lymphocytes, and inversion in proportional representation of memory versus naïve cells (T memory cells increase and T naïve cells decrease with age). Altered immunoregulation results in an increase in autoimmune syndromes (SLE, RA, SS, others), oligoclonal expansion of T− and B−cells (decrease in diversity with age), as well as an increase in monoclonal gammopathies.
It has been suggested that while the process of replicative senescence is applicable to human immune cells, it is not relevant to immunological aging in murine models.(98) This formulation is based on the facts that (1) given the relatively longer telomere length and greater telomerase activity in mice, telomere shortening after T−cell activation is likely to have a greater impact on the regulation of CD 8 T−cells in humans, especially with their longer life span, and (2) CD28 expression, a potent co−stimulator required for optimal telomerase induction in antigen−specific T cells, is lost by the CD8 pool with age and replicative senescence in humans but not in mice.(99)
Replicative senescence of CD8 T−cells is also associated with the reduced ability to produce interferon−gamma (IFγ), potentially influencing the complex regulation of bone remodeling in favor of osteoclastic resorption.(98) It has also been suggested that increased production of tumor necrosis factor−alpha (TNFα) and interleukin−6 (IL−6) by senescent CD8 T−cells could promote osteoclast maturation and activation, influencing age−related bone alterations.(100) For example, increased proportions of CD8 T−cells expressing senescence−related surface markers have been correlated with osteoporotic fractures in older women.(101)
Emerging Concepts
The idea that intrinsic cell aging leads to age−related tissue dysfunction and organismal decline directly from exhausted replicative capacity has given way to the notion that cell senescence promotes a variety of deleterious effects independent of removing cells from the proliferative pool. For example, stress resistance correlates strongly with longevity, and replicative capacity in culture may be an indirect measure of cellular sensitivity to stress. Senescence−associated growth arrest is also accompanied by changes in cell phenotype, including the differential expression of secreted proteins that may facilitate age−related pathology such as macular degeneration and cancer promotion/progression. By virtue of their growth arrested state, gradual accumulation of senescent cells may in part also explain some age−related losses in tissue structure and function.
Mechanistically, the roles of polycomb group complexes (PcGs) and stress−activated p38/MAP kinases are emerging as contributors to the senescence−associated upregulation of p16.(42)(102)(103) Small, noncoding RNAs or micro RNAs, especially those of the Mir−34 family, are activated by p53 and have been implicated in the senescence program.(104) So−called FOXO proteins are thought to inhibit cell growth by a direct effect on proliferation−related genes, and/or by indirect effects in response to metabolic changes, stress and even cell recycling/remodeling (autophagy).(105)(106)(107) Autophagy is increased in senescent cells and may also play a role in tumor suppression.(108)(109).
Cellular senescence can be initiated through multiple mechanisms to achieve the hallmark of essentially irreversible growth arrest, and there appears to be multiple pathways by which senescent cells have functional consequences at the level of tissue and perhaps organismal aging.