Course Authors
Arlene Tan Tieng, M.D., Irene Blanco, M.D., and Peter Barland, M.D.
Release Date: 08/10/2009
Upon completion of this Cyberounds®, you should be able to:
Define the hemophagocytic syndrome (HPS)
List the rheumatic diseases that have been associated with reactive HPS
Describe the clinical and lab manifestations of this disorder
Discuss the life-saving treatments that should be considered early when autoimmune-associated hemophagocytic syndrome (AAHS) is suspected.
|
Case Report of a Patient with Hemophagocytic Syndrome. AH, a 23-year-old male, was admitted to our institution with five days of fever, abdominal pain and non-bloody diarrhea. He was febrile to 101.2°F, and his exam revealed a benign abdomen and no rash, lymphadenopathy or synovitis. He was found to be pancytopenic with a WBC of 2600/mm3, an absolute neutrophil count of 1700/mm3, a hemoglobin of 8.9 gm/dL and platelets of 22,000/mm3 with schistocytes seen on the peripheral blood smear. His International Normalized Ratio (INR) was increased at 1.6, the alanine aminotransferase (ALT) was elevated to 87 U/L and ferritin was 6042 mg/L. He was started on empiric antibiotics with pipercillin/tazobactam for possible overwhelming infection causing bone marrow suppression. A work-up for acute bacterial infection, viral infection including CMV, HBV, Parvovirus, viral hepatitis, HIV, as well as Ehrlichiosis and Babesiosis, was negative. The INR reverted to normal after vitamin K was administered and a bone marrow biopsy was scheduled to evaluate the possibility of acute leukemia, lymphoma or myelodysplastic syndrome. However, AH's family signed him out of the hospital against medical advice. |
One month later AH returned complaining of fever, vomiting and diarrhea. He was febrile to 104.0°F. As on his previous admission, he was found to be pancytopenic with 3-4 schistocytes per hpf and his ferritin was elevated to 28,583 mg/L. A CT of the abdomen and pelvis showed diffuse lymphadenopathy and splenomegaly. A bone marrow aspirate and biopsy were unremarkable. A lymph node biopsy showed extensive hyalinization with no evidence of lymphoma.
Given that AH remained pancytopenic, had fevers and elevated ferritin levels, the diagnosis of hemophagocytic syndrome was entertained. A repeat bone marrow biopsy showed increased monocytes/histiocytes accounting for 10-15% of the bone marrow that was confirmatory of a diagnosis of primary hemophagocytic syndrome. AH improved with the administration of solumedrol 100 mg IV QD, becoming afebrile with decreased ferritin levels, and normalization of haptoglobin and fibrinogen. However, he remained pancytopenic.
The decision was made to start treatment for hemophagocytic syndrome using dexamethasone 10 mg/m2, cyclosporine 4 mg/kg and etopside twice weekly at an initial dose of 75 mg/m2, as well as IVIG 0,5 mg/kg every four weeks. AH received one dose of etoposide; however, further immunosuppression was held due to bacteremia with vancomycin resistant enterococcus. AH was continued on the dexamethasone and was treated with daptomycin for 10 days. Etoposide was then restarted and cyclosporine was started at the appropriate dose. However two days after the administration of etoposide and cyclosporine, AH became lethargic, hypoxic and hypotensive. He was admitted to the ICU with presumed septic shock; however, soon after admission to the ICU, he went into pulseless electrical activity (PEA) and arrest. Resuscitative efforts failed.
Discussion
The hemophagocytic syndrome (HPS), also known as hemophagocytic lymphohistiocytosis (HLH), is characterized pathologically by systemic proliferation of morphologically benign histiocytes engaged in abnormal phagocytosis of hematopoietic cells. These non-neoplastic histocytes infiltrate the bone marrow, spleen, liver or lymph nodes. Clinical and laboratory features include fever, hepatosplenomegaly, lymphadenopathy, severe cytopenias, and elevated levels of serum ferritin, triglycerides and hepatic enzymes. HPS is a rare but potentially life-threatening complication of Still's disease in both children and adults, of systemic lupus erythematosus and rarely with other systemic autoimmune connective tissue diseases. In addition to its occurrence in connective tissue diseases, HPS may be secondary to infections, malignancies and drugs. When the syndrome occurs in these settings it is referred to as secondary, acquired or reactive HPS. The term macrophage activation syndrome (MAS) has also been used instead of reactive HPS.(1)
In addition, HPS can occur as a primary, or genetic, disorder. The primary form can be further classified into familial hemophagocytic lymphohistiocytosis (FHL) and the immune deficiencies of X-linked lympho-proliferative syndrome (XLP), Chédiak-Higashi syndrome (CHS) and Griscelli syndrome (GS) The common finding in all of these primary forms of HPS has been a defect in cytotoxic lymphocyte function. In this Cyberounds® conference we review the key clinical, biochemical and pathological features of this syndrome with special attention to reactive HPS occurring in association with autoimmune disorders. Recognition of these features makes it possible to diagnose HPS early. We also review possible pathogenetic mechanisms for the syndrome and discuss its current management.
Background
Scott and Rob-Smith first described HPS in 1939 as histiocytic medullary reticulocytosis, a disorder characterized by fever, pancytopenia, hepatosplenomegaly, and lymphadenopathy due to infiltration by phagocytizing histiocytes.(2) Subsequent to their original report that occurred in a patient with a malignancy, others have described the same histopathologic and clinical picture in association with viral and bacterial infections, and lymphoma.(3) The first description of HPS occurring in association with active systemic lupus erythematosus (SLE) was made in 1991 by Wong et al., who reported six patients whose clinicopathological features included fever, pancytopenia, marrow hemophagocytosis, hypocomplementemia, high antinuclear antibody titer, and cutaneous and visceral vasculitis.(5) The HPS occurred in association with a flare of SLE. In these cases, the pancytopenia responded to corticosteroid therapy. In 1997, Kumakura et al. reported patients with mixed connective tissue disease (MCTD) and progressive systemic sclerosis (PSS) who demonstrated reactive HPS.(6) In 2000, García Escudero et al. reported HPS in Churg-Strauss vasculitis.(7) In 2003, Dhote et al. reported 26 cases of reactive HPS occurring in the course of several systemic autoimmune diseases including SLE, rheumatoid arthritis (RA), adult Still's disease (ASD), polyarteritis nodosa, mixed connective tissue disease, systemic sclerosis and Sjögren's syndrome.(8) Finally in 2008, Fukaya et al. reported HPS in a series that included patients with polymyositis, dermatomyositis and microscopic polyangiitis.(9) Of the rheumatic diseases, the conditions most frequently associated with reactive HPS are SLE and especially ASD, with a prevalence of 0.9-2.4% and 12%, respectively.(9)
Diagnosis
Henter et al. presented the first set of diagnostic guidelines for HPS, which included five criteria: fever, splenomegaly, cytopenias affecting at least two lines in the peripheral blood (hemoglobin ≤9 g/dL, platelets <100,000/μL, neutrophils <1000/μL), hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis in bone marrow, spleen or lymph nodes.(10) If hemophagocytic activity is not proven at the time of presentation, further search for hemophagocytic activity is encouraged. If the bone marrow specimen is not conclusive, material may be obtained from other organs. Serial marrow aspirates over time may also be helpful. Hemophagocytosis can also be seen in other body fluids, such as peripheral blood, pleural effusions, cerebrospinal fluid, urine and ascitic fluid.(11) Henter's original guidelines have been expanded to include three additional criteria: low or absent natural killer (NK)-cell activity, hyperferritinemia, and high levels of soluble interleukin (IL) -2 receptor.(12) Five of these eight criteria need to be present to establish the diagnosis.
Other abnormal clinical and laboratory findings consistent with the diagnosis are cerebromeningeal symptoms, lymph node enlargement, jaundice, edema and skin rash. Laboratory findings commonly found in HPS include hepatic enzyme abnormalities, hypoproteinemia, hyponatremia, VLDL↑, HDL↓.

Figure 1. Bone Marrow Aspirate Showing Hemophagocytic Macrophage.
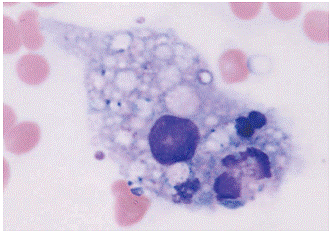
Source: Katoh N, Gono T, Mitsuhashi S, Fukushima K, Takei Y, Matsuda M, Ikeda S. Hemophagocytic syndrome associated with rheumatoid arthritis. Intern Med. 2007; 46(21):1809-13. Epub 2007 Nov 1. Courtesy of Japanese Society of Internal Medicine.
Given that HPS may be secondary to infections, malignancies and drugs, as well as systemic autoimmune diseases, the aforementioned features may not be entirely applicable to autoimmune disease. Kumakura et al. (13) proposed the following diagnostic criteria for autoimmune-associated hemophagocytic syndrome (AAHS):
- Cytopenia (affecting ≤2 of 3 lineages in the peripheral blood and not caused by an aplastic or dysplastic bone marrow).
- Histiocytic hemophagocytosis in bone marrow or other reticuloendothelial systems including spleen, liver or lymph nodes.
- Active phase of underlying autoimmune disease at the occurrence of hemophagocytosis.
- Other reactive hemophagocytic syndromes such as virus- or malignancy-associated hemophagocytic syndrome are excludable.
One of the most impressive findings in reactive HPS is the presence of very elevated levels of serum ferritin with levels frequently exceeding 50,000 ug/l. Ferritin is located in all cells, but largely in hepatocytes. When ferritin is secreted from tissue into plasma, carbohydrate residues are added. The usual ratio of glycosylated to non-glycosylated ferritin in serum is 50-80%. In reactive HPS, whether the underlying cause is autoimmune disease, infection, neoplasm or inflammation, the percentage of glycosylated ferritin is particularly low (i.e., <20%). The explanation for the diminished glygosylation of ferritin remains unknown but several hypotheses have been proposed: hepatic function alteration, massive cell lysis, an incomplete glycosylation process and active down-regulation of the glycosylation process. The percentage of glycosylated ferritin may be a helpful marker for the diagnosis of reactive HPS.(14)(15))
An increased concentration of urinary β2-microglobulin (β2m) is another marker for reactive HPS. In Japan, SLE and RA patients with reactive HPS manifested remarkably increased urine levels of β2m, which normalized when the reactive HPS improved with treatment. The levels of urinary β2m appear to correlate with levels of serum interferon-gamma (IFN-γ) -- an inflammatory cytokine that has been thought to activate the macrophages in reactive HPS. The urinary β2m may be an additional lab marker to diagnose HPS associated with collagen vascular diseases.(16)
Pathogenesis
The pathophysiological basis for HPS is not fully understood. A characteristic finding in HPS appears to be the impaired function of natural killer (NK) cells of the innate immune response and cytotoxic T cells of the adaptive immune response. Both NK cells and cytotoxic T cells use the same mechanism of killing. HPS appears to be caused by the impaired function of NK cells and cytotoxic T cells, which leads to the accumulation of stimulated macrophages that phagocytose circulating blood cells and to the production of inflammatory cytokines. Natural killer cells and cytotoxic T cells of innate and adaptive immunity, respectively, use the same mechanism of killing. These cells secrete specialized cytotoxic granules that contain cytotoxic effector proteins, which penetrate the cell membrane of the target cell and trigger apoptosis.(17) A number of mutations in the formation and function of the cytoxic granules have been identified in patients with familial forms of HPS. One of the important functions of the cytotoxic lymphocytes is the down regulation of macrophages which have been activated during innate and adaptive immune responses such as occurs with systemic infections and autoimmune diseases. These activated macrophages are phagocytic and secrete proinflammatory cytokines. It is believed that in HPS unregulated macrophages phagocytose circulating blood cells, and promote the infiltration of activated lymphocytes. Consistent with this proposed pathogenic mechanism are the observations that patients with active HPS have significantly raised serum levels of the TH1 lymphocyte cytokines IFN-γ, IL-12, and IL-18, compared with those in remission or healthy controls.(18) In addition, serum levels of the pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α), IL-1β and IL-6 are also higher than in controls.
Treatment
There is currently no standard treatment protocol for reactive HPS in adults. Given the heterogeneity in the etiology of HPS, therapy has typically been individualized in each unique case. In a series of 30 cases, the mortality rate of solely autoimmune-associated HPS without any concurrent infection was reported to be 5.3%. The inclusion of infection in this case series yielded an overall mortality rate of 20%.(9) Thus, treatment for HPS should begin promptly before the development of infection secondary to the leukopenia. Corticosteroids are first-line therapy, and granulocyte colony stimulating factor (G-CSF) should be considered for neutropenia.(18) Infection control is also priority, as HPS can be caused by as well as complicated by a viral, bacterial, fungal or parasitic infections. If there is no evidence of an active neoplasm and if the ferritin has peaked within the past two days, then giving intravenous immunoglobulin (IVIG) 1 g/kg over two days has shown improvement without much toxic effects.
In autoimmune-associated reactive HPS, a relatively good response is achieved with corticosteroids. High doses of corticosteroids are cytotoxic for lymphocytes and inhibit expression of inflammatory cytokines. Methylprednisolone pulse therapy (1 g/day for 3 days), half-dose of pulse therapy (0.5 g/day for 3 days), and high-dose corticosteroid therapy (prednisolone 1 mg/kg/day) have been reported to result in improvement.(14) In corticosteroid-resistant autoimmune associated HPS, Fukaya and colleagues recommend to start cyclosporine (CsA) early.(9) CsA prevents T-lymphocyte activation. One dose suggested is CsA 2 to 5 mg/kg per day, renal-dosed when necessary. Although CsA and intravenous cyclophosphamide pulse therapy (IVCY) 500-700 mg/day demonstrated similar efficacy, CsA is preferred, as the latter is more likely to lead to bone marrow suppression.(14)
In addition to these conventional therapies for reactive HPS, there have been some other newer and surprisingly successful treatments. In SLE, for instance, TNF blockade is typically avoided as it has been associated with the production of autoantibodies against anti-dsDNA and anticardiolipin, which could provoke flare and thrombosis, respectively. Nonetheless, the TNF-α inhibitors have been used successfully in lupus-associated HPS. Infliximab (5mg/kg IV every 6 to 8 weeks) and etanercept (25 mg, twice a week) were shown to be effective therapies in SLE patients with HPS who were refractory to high-dose steroids, CsA and IVIG.(19)(20) It must be noted, though, that some antirheumatic drugs have induced HPS. These include non-steroidal antiinflammmatory drugs, gold, bucillamine, sulfasalazine, methotrexate and etanercept.(16)(21)
Conclusions
Since its initial characterization in the 1930s, HPS has been observed in infection, malignancy, drugs, and only beginning in the 1990s, rheumatic diseases. Diagnostic criteria for AAHS consist of at least two cytopenias during the active phase of an autoimmune disease and hemophagocytosis in the reticuloendothelial system. In spite of this, it can be difficult to confirm hemophagocytosis on biopsy or aspirate. Other characteristics of HPS include fever, splenomegaly, hypertriglyceridemia, hypofibrinogenemia and hyperferritinemia. Some of these features occur late in the disease course, and it is not essential to fulfill all criteria before starting therapy. It is of crucial importance for clinicians to consider HPS in rheumatic diseases early, as treatment can be life-saving.
Physicians can request treatment protocols for HPS by completing a form from this website:
www.HistiocyteSociety.org