Course Authors
Robert Foronjy, M.D., and Jeanine D'Armiento, M.D., Ph.D.
Release Date: 06/29/2009
Upon completion of this Cyberounds®, you should be able to:
List the pathologic features of chronic bronchitis and emphysema
Discuss whether MMPs contribute to the development of COPD
Discuss if MMP inhibitors might effectively treat or prevent COPD.
Chronic obstructive pulmonary disease(COPD) is one of the leading causes of death in the United States. Twenty-four million Americans are affected with this condition and in 1998 it accounted for 110,000 deaths nationwide.(1)(2) As the population ages and a new generation of smokers matures, it is certain that this disease will impact patients and the medical establishment for years to come.
The defining feature of COPD is irreversible airflow limitation during forced expiration. This may result from loss of elastic recoil as a consequence of lung tissue destruction or from an increase in the resistance of the conducting airways. The severity of this airflow limitation is measured by the forced expiratory volume in 1 second (FEV1) and its ratio to forced vital capacity (FVC), FEV1/FVC.
Though COPD is a heterogeneous disease, it has been stratified into two general categories: chronic bronchitis and emphysema. Chronic bronchitis is characterized by hyperplasia of mucus producing glands that results in airway obstruction secondary to excessive mucus production. The pulmonary vascular bed remains relatively intact and cardiac output is preserved or increased. The pathophysiology of chronic bronchitis -- a deficit in ventilation compared to perfusion (i.e., V/Q mismatch) -- produces hypoxia and polycythemia. In contrast, emphysema is characterized by destruction of the airspaces distal to the terminal bronchioles in the lung. (3) Unlike chronic bronchitis, this process results in the loss of the pulmonary capillary bed as well as alveolar tissue. Due to the absence of alveolar-capillary units, oxygenation is impaired. However, overall oxygen levels in the blood are preserved by hyperventilation.
Chronic Bronchitis and MMPs
Though the pathophysiology of chronic bronchitis and emphysema is distinct, there are common processes that are linked to their development. For one, both syndromes are caused by chronic exposure to cigarette smoke, which generates a robust inflammatory response in the lung that results in a marked increase in proteases within the lung.(4) Animal and human studies indicate that the presence of these proteases is a critical factor in the development of these diseases.(5)(6)
In chronic bronchitis, the elaboration of matrix metalloproteinases (MMPs), zinc-dependent proteases, within the airways contributes to the disease through several mechanisms. Neutrophilic inflammation within the airway is a hallmark of chronic bronchitis and both MMP-9 and MMP-12 have been demonstrated to mediate this inflammatory reaction.(7) MMP-9 cleaves CXCL8 (IL-8) increasing its chemotactic activity for neutrophils 10-fold(8) and proteolytic processing by MMP-9 can activate pro inflammatory cytokines such as IL-1β,(9) TGF-β(10) and TNF-α.(11) In animal models, the loss of MMP-12 decreases cigarette smoke-induced inflammation in the lung,(12) while instillation of recombinant MMP-12 into the airways of mice causes neutrophilic inflammation and gelatinase activation.(13) Both MMP-9 and MMP-12 cleave elastin which generates degradation peptides that drive disease progression through their chemotactic effects.(14)
In addition to inflammation, the MMPs have been linked to goblet cell hyperplasia(15) and the induction of mucin expression within the lung epithelium.(16) Furthermore, MMPs stimulate dysfunctional airway remodeling that exacerbates the obstruction that is caused by inflammation and mucus production.(17) Increases in MMP-9(18) and MMP-12(19) have been detected within the lungs of chronic bronchitis patients, and the levels of MMP-9 correlate well with the degree of lung impairment as measured by FEV1.(18) Inhaled steroids, which are so effective at treating airway inflammation in asthma, have no impact on the levels of MMPs isolated from the lungs of COPD patients.(20) The inability to treat this proteolytic component is believed to be an important factor in the progression of the disease.(21) Together, these studies indicate that the presence of MMPs within the airways of smokers is a central factor in the development of the pathophysiologic changes that occur in chronic bronchitis.
Emphysema
Emphysema is an anatomic disease(22) characterized by the loss of alveolar tissue with resultant physiologic changes that impair the respiratory function of the patient.(23) While 90% of emphysema patients are smokers, only 10-15% of smokers will actually develop the disease. The variable incidence of this disease among smokers points out that genetic factors are an important determinant of the development of the disease. While an inherited deficiency of alpha-1-anti-trypsin increases the risk of emphysema formation, less than 1% of emphysema patients demonstrate this deficiency. This indicates that other, yet to be determined, genetic factors alter the risk of disease development. Indeed, the precise mechanisms responsible for the destructive changes that occur in this disease have not been fully determined.
Aside from smoking, other environmental factors such as pollution, toxic chemical exposure, and chronic infections significantly influence disease development. In fact, approximately 10% of all COPD patients are non-smokers.(24) While chronic bronchitis affects the more central airways, the peripheral airways undergo distinct changes in emphysema. Indeed, marked subepithelial and adventitial fibrosis are noted in the small airways of emphysema patients and these changes are strongly associated with disease progression.(25) Studies have shown that cigarette smoke directly induced profibrotic growth factors and collagen synthesis in the small airways of mice,(26) and this remodeling can alter lung mechanics and contribute to disease progression.(27)
The Role of Elastin and Elastases in Emphysema
Given the structural changes that occur in emphysema, researchers have focused on the role of the extracellular matrix.(28) The extracellular matrix is comprised of structural proteins such as collagen and elastin that maintain the architecture of the lung, thus allowing it to fulfill its function of exchanging oxygen and carbon dioxide with the circulation. Disruption of this matrix has been recognized to be a central factor in the development of emphysema.(29) However, the exact mechanisms that lead to the structural changes in the lung are a matter of continuing debate.
Significant evidence has implicated elastin in the pathogenesis of this disease,(30) as the disruption of elastin exerts a pivotal role in disease development.(31) Elastin is a key component of the lung extracellular matrix that provides the tissue the resiliency to tolerate repeated mechanical stress.(32)(33) The importance of elastin is underscored by the fact that smokers with a hereditary deficiency of α1-AT (the main inhibitor of neutrophil elastase) are predisposed to the development of early-onset emphysema.(34) Furthermore, the intra-tracheal administration of elastase -- but not other proteases -- produces emphysematous changes in experimental animal models;(35) electron microscopy analyses of human emphysema reveals disruption and disorganization of elastin fibril sheets in the lung.(36) These studies provide significant affirmative evidence for the importance of elastin and elastolytic enzymes in this disease process.
The Effect of MMPs on Lung Elastin in Emphysema
Several studies have implicated the elastolytic proteases MMP-9 and MMP-12 in the pathogenesis of emphysema. MMP-9 is a potent elastase that on a molar ratio is 30% as effective as human leukocyte elastase at solubilizing elastin.(37) Alveolar macrophages from smokers release greater amounts of elastases compared to control macrophages(38) and this activity is largely due to MMPs that are expressed in the cell,(39) specifically MMP-9 and MMP12. Indeed, macrophages from COPD patients have increased MMP-9 expression(40) and release greater amounts of MMP-9 upon stimulation with cigarette smoke compared to macrophages from smoking and non-smoking control subjects.(41) Increased MMP-9 levels were detected in emphysema lung samples(42) and enhanced expression of this protease was noted in alveolar macrophages from COPD patients.(41)(43) Similarly, elevated MMP-9 expression and activity were identified in animal models of the disease(44)(45) and were thought to play a role in the destructive process. In addition, patients with a polymorphism that increases MMP-9 promoter activity were at increased risk of developing emphysema.(46)
Our laboratory demonstrated a role for MMP-9 in lung destruction through the generation of transgenic mice that expressed human MMP-9 within tissue macrophages. These mice developed progressive air space enlargement in the lung, coupled with a marked decrease in elastin within the alveolar walls of the mice. This is significant as elastin and type III collagen fibrils are key structural elements of the alveolar wall.(47) Thus, this study provides direct evidence that MMP-9 generates emphysematous changes by degrading elastin in the alveolar wall. Recently, it was shown that mice deficient in MMP-12, a major elastolytic protease, were resistant to the development of smoke-induced emphysema.(48) Elevated MMP-12 levels were detected in the macrophages(49) and epithelial cells(50) from COPD patients. Furthermore, inhibition of the elastolytic proteases MMP-9 and MMP-12 delayed the development of cigarette smoke-induced emphysema in guinea pigs.(51) Thus, these studies demonstrate that the degradation of elastin by MMPs is a pivotal process in emphysema formation.
The Role of MMPs on Lung Collagen in Emphysema
In the past decade, studies have emerged suggesting that collagen and collagen-degrading enzymes are key elements in the pathogenesis of emphysema. In a transgenic mouse line with lung-specific expression of MMP-1, morphometric evidence of emphysema developed that was pathologically similar to that seen in the human disease.(5) Given that MMP-1 has activity primarily against fibrillar collagens (collagens type I, II and III) and not elastin, this study demonstrated that emphysema could develop via an elastin-independent mechanism. Subsequently, studies in guinea pigs showed that cigarette smoke exposure increased expression of collagenase and generated morphometric evidence of collagen breakdown and repair within the lung.(52)(53)
Studies in humans have also demonstrated the presence of collagenase in the lung and macrophages from emphysema patients. Finlay and colleagues showed that alveolar macrophages from the bronchoalveolar lavage of emphysema patients had augmented production of collagenase compared to matched controls.(43) Ohnishi et al. reported a significant increase in collagenolytic activity within the lung parenchyma of emphysema patients.(42) Our laboratory demonstrated the presence of MMP-1 mRNA, protein and collagenase activity within the lung parenchyma of patients with emphysema compared to normal controls.(6) In this study, lung samples were examined from 23 patients with emphysema undergoing lung reduction surgery or lung transplantation. When immunolocalization studies were performed on these samples, the type II pneumocyte was identified as the cell type producing collagenase.(6) These studies were the first to demonstrate that destructive enzymes in emphysema were coming not from the inflammatory cell population but the innate lung epithelial cells. This has led to a reexamination of the pathogenesis of the disease and a greater focus on the fundamental changes occurring within the lung parenchyma post smoke exposure.
The Importance of Collagen Subtypes in Emphysema
Transgenic mice with a collagenase resistant Type I collagen were utilized to demonstrate that the expression of MMP-1 in mice causes the selective loss of type III collagen within the alveolar region of the lung.(47) The loss of type III collagen was associated with emphysematous changes coupled with an increase in lung compliance.(47) The findings suggest that changes in the distribution of collagen subtypes within the lung may have significant structural and physiological effects on the lung even in the presence of unchanged levels of total collagen and elastin. Collagen comprises 60% of total lung protein with approximately 60% being type I collagen and 20-30% type III collagen.(54)
Despite the fact that collagen constitutes a significant portion of the extracellular matrix, the exact contribution of type III collagen to the biomechanical properties of the lung is not yet fully understood.(55)(56) The fibrils of type III collagen are a significant component of the alveolar wall(57) and are found in intimate associate with elastin fibrils in this region.(58) These two structural elements are believed to work synergistically to contribute to the extensibility of lung tissue. The compound transgenic studies described above(47) further add to the understanding of the function of the type III collagen fibrils by demonstrating that their selective loss from the alveolar walls leads to airspace dilation and increased pulmonary compliance. This is also supported by the fact that patients with Type VI Ehlers-Danlos syndrome (a deficiency of type III collagen) develop emphysema and increased lung compliance.(59)(60)(61)
Changes in lung collagen have been demonstrated in several experimental models of emphysema. Increased hydroxyproline levels are seen in the lung of mice exposed to cigarette smoke.(62) Intratracheal elastase causes an increase in collagen deposition and aberrant collagen remodeling(36) in rats and, in hamsters, leads to an increase in the transcription of type I collagen coinciding with the development of emphysema.(63) Human studies also demonstrate that the overall collagen content is increased in the lung tissue from patients with emphysema.(64)(65)
How then is the finding of increased collagen consistent with the fact that collagenolysis leads to emphysema? One possible mechanism is that MMP-1 alters the normal turnover of collagen in the lung leading to a decrease in the content of type III collagen. If the total collagen content is unchanged and the ratio of type III/I collagen is decreased, there would be a net increase in type I collagen. Transgenic mouse studies have shown that expression of MMP-1 directly stimulates the transcription of Type I collagen.(66) This suggests that the lung in emphysema is comprised of a remodeling tissue with loss of type III collagen and deposition of type I collagen. The alteration of the collagen subtypes and the overall remodeling that occurs may change the mechanical properties of the lung and lead to the pathology observed.(67) Further studies are warranted to evaluate the role of these collagen subtypes in the pathogenesis of the disease.
MMPs and Extracellular Matrix in Emphysema
It is likely that cigarette smoke mediates its destructive changes through both alterations in collagen and elastin. The disease model set forth is outlined in Figure 1. Cigarette smoke leads to the recruitment of macrophages into the lung. These macrophages secrete cytokines which further augment the inflammatory response, leading to the induction and release of proteolytic enzymes by macrophages and neutrophils. This proteolytic cascade leads to both collagen and elastin degradation. The macrophages and cigarette smoke then stimulate the parenchymal lung cells in patients susceptible to emphysema to produce proteolytic enzymes. It is hypothesized that the induction of proteolytic enzymes within the epithelial cells of the lung results in an irreversible state of lung damage and repair leading to the progressive damage seen in emphysema.

Figure 1. Smoking-Induced Lung Damage Model.
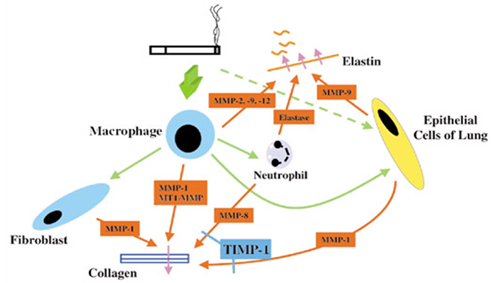
After we determined that parenchymal epithelial cells produce significant amounts of MMP-1, our laboratory demonstrated through in vitro and in vivo studies that cigarette smoke, the major etiological factor in emphysema, can directly induce MMP production in epithelial cells in a MAP Kinase dependent fashion.(68) To confirm relevance to the human disease, we obtained human lung samples from patients with emphysema to examine if changes in phosphorylated extracellular regulated kinase (p-ERK) were associated with disease. When specific p-ERK activity was examined, significantly higher levels were detected in lung homogenates from patients with emphysema, compared to normal controls (1.9 ± 0.2 fold increase above normal lung, P=0.02). The identification of the signaling pathway altered by cigarette smoke in vitro and in vivo will allow the identification of novel therapeutic targets in the disease.(68) Ongoing studies in our laboratory are investigating the use of several MAP kinase inhibitors as therapeutic agents in chronic obstructive pulmonary disease (COPD).
In subsequent studies, to determine whether the MMP-1 promoter was directly activated by cigarette smoke exposure, and to identify the specific transcriptional element(s) responsible for MMP-1 mRNA induction, a series of promoter deletions studies were performed.(69) These deletion studies led to the identification of a novel cigarette smoke responsive (CSR) element present within the promoter region of both MMP-1 and identified a unique role for the MAP Kinase pathway in this disease.(69) Further dissection of the signaling pathway altered by cigarette smoke exposure will allow us to better understand the molecular changes that occur within the lungs of patients with emphysema. It will also help us elucidate the downstream effects of cigarette smoke and the resulting tissue damage that occurs as a result of cigarette smoke exposure. This information is useful not only in emphysema but in cardiovascular diseases caused by chronic smoke exposure and can potentially lead to the identification of novel therapeutic targets in these diseases. In addition, the identification of a cigarette responsive element in the MMP-1 promoter allows us to pursue genetic studies to examine if polymorphisms in this region of the promoter are responsible for susceptibility or severity of disease.
MMP inhibitors to Treat or Prevent Disease
MMP activity within the tissue is controlled at several steps, which include gene expression of MMPs, production (transcription and secretion) of MMPs, activation of proMMPs and inhibition of MMP activities by natural inhibitors (TIMPs and α2-macroglobulin). In addition, the activation processes of proMMPs have been intensively studied and the accumulated data indicate that proMMPs are activated by various mechanisms.(70) Thus, these are potential targets for the regulation of MMPs but inhibition by synthetic low molecular weight inhibitors to MMPs is more realistic for the treatment of disease.
For the past 30 years, over 56 matrix metalloproteinase (MMP) inhibitors have been pursued as clinical candidates in various therapeutic areas, mainly for targeting cancer, arthritis, or cardiovascular diseases.(71) Despite these efforts, the majority of the inhibitors have failed in clinical trials for various reasons including musculo-skeletal problems such as arthralgia, myalgia and tendonitis, and other negative side effects of the nonspecific MMP inhibitors.(72)(73). Thus, no MMP inhibitor compound has been licensed to date.(74) The most challenging aspect of developing MMP inhibitors is finding candidates having acceptable pharmacological parameters including pharmokinetic and selectivity profiles.(75) Because of the availability of crystal structures for many MMPs,(76) the second-stage MMP inhibitors with more selectivity to MMP species have been developed, and they appear to be less toxic.
Treatment of smoke-exposed guinea pigs with one of these inhibitors, an MMP-9 inhibitor (CP-471 and CP-474) reduced inflammation and attenuated the initiation of lung destruction; however, after 4 months of smoke exposure the extent of emphysema was not different between treated and untreated smoke-exposed guinea pigs.(77) This inhibitor did not have activity against the collagenases.(77) It is not clear which are the relevant MMPs to inhibit in emphysema and the target enzymes may be different at different stages of the pathophysiological process. However, an intriguing possibility is that collagenolytic enzymes affect matrix remodeling in the late stages of emphysema and are responsible for the progressive tissue destruction.
Further studies using experimental animal models are needed to develop effective strategies for treatment of COPD. The mouse and guinea pig models of smoke induced emphysema do not express MMP-1, a major destructive enzyme in humans. Therefore, rabbits or higher level species are required to adequately test these therapies. MMP inhibitors specific to a single critical MMP or combined inhibitors that block critical MMPs will likely be required to treat emphysema. An advantage in the treatment of lung disease, however, is the ability to develop aerosolized compounds that may limit the side effects seen in the systemic delivery of compound.