Course Authors
Kelly A. Diggs-Andrews, B.S., Julie M. Silverstein, M.D., and Simon J. Fisher, M.D., Ph.D.
Ms. Diggs-Andrews is currently a senior Ph.D. student in the Molecular Cellular Biology program in the Division of Biology and Biomedical Sciences at Washington University in St. Louis; Dr. Silverstein is currently a clinical fellow and Dr. Fisher is currently an Assistant Professor of Medicine, Cell Biology & Physiology, both in the Division of Endocrinology, Metabolism & Lipid Research, Washington University, St. Louis, MO.
Within the past 12 months, Ms. Diggs-Andrews and Dr. Silverstein report no commercial conflicts of interest; Dr. Fisher has been a member of the Speakers Bureau for Merck. This relationship will not influence his presentation.
This activity is made possible an unrestricted educational grant from  .
.
Estimated course time: 1 hour(s).

Albert Einstein College of Medicine – Montefiore Medical Center designates this enduring material activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
In support of improving patient care, this activity has been planned and implemented by Albert Einstein College of Medicine-Montefiore Medical Center and InterMDnet. Albert Einstein College of Medicine – Montefiore Medical Center is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Upon completion of this Cyberounds®, you should be able to:
Identify the sites in the brain that sense and respond to changes in blood sugar
Identify some of the key glucose sensing proteins in the brain
Describe the hierarchy of hormones that are released in response to hypoglycemia
Discuss the etiology of the impaired counterregulatory response and hypoglycemia unawareness that occurs in people with diabetes
List some therapeutic interventions shown to reduce the risk of hypoglycemia.
The brain is an obligate glucose consumer and critically dependent on glucose supply for normal function. Maintaining blood glucose levels within a tight physiological range is critical to whole-body glucose homeostasis. For patients with diabetes, many of the glucose lowering drugs used to treat hyperglycemia can reduce blood sugar below the physiological range and induce a state of hypoglycemia (low blood sugar). In response to hypoglycemia, as a means of self-preservation, the brain coordinates a stress (counterregulatory) response to rapidly restore blood sugar levels to normal. This counterregulatory response involves systemic hormone responses, including a reduction in insulin secretion from the pancreatic b-cells and an increase in glucagon secretion from the pancreatic a-cells.
The counterregulatory stress response also involves activation of critically important glucose sensing areas in the hypothalamus which respond to hypoglycemia by activating sympathetic efferent signals to the adrenal medulla to rapidly release epinephrine. The hypothalamic release of corticotrophin releasing hormone (CRH) mediates release of adrenocorticotrophin hormone (ACTH) systemically to trigger cortisol release from the adrenal cortex, which may be an important counterregulatory response to more prolonged hypoglycemia (Figure 1).
Abbreviations: CNS, central nervous system; SNS, sympathetic nervous system; CRH, corticotrophin releasing hormone; ACTH, adrenocorticotropin hormone; HPA, hypothalamic-pituitary-adrenal; ARC, arcuate nucleus of the hypothalamus; PVN, paraventricular nucleus of the hypothalamus; VMH, ventromedial hypothalamus; ICV, intracerebroventricular; GLUT, glucose transporter; GI, glucose-inhibited neuron; GE, glucose-excited neuron; CGM, continuous glucose monitor; KATP , potassium dependent ATP channel; GK, glucokinase; AMPK, adenosine monophosphate activated protein kinase; CSII, continuous subcutaneous insulin infusion; MDI, multiple daily injections; HAAF, hypoglycemia associated autonomic failure.

Figure 1. Glucose Sensing and the Physiological Response to Hypoglycemia.
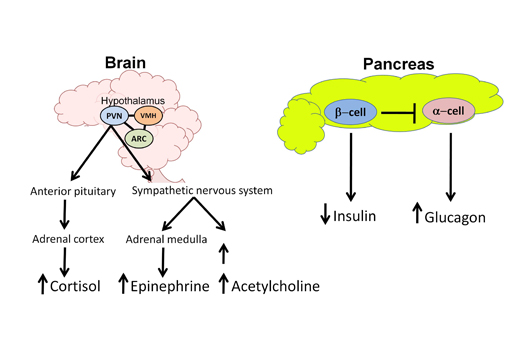
In the normal response to hypoglycemia, the brain (especially the hypothalamus) coordinates and triggers a stress response to restore euglycemia. Key areas of the hypothalamus that sense and respond to hypoglycemia are the ventromedial hypothalamus (VMH), the arcuate nucleus (ARC) and the paraventricular nucleus (PVN). Efferent outflow from the hypothalamus results in activation of the hypothalamic-pituitary-adrenal (HPA) axis as well as the sympathetic nervous system to increase production of key counterregulatory hormones. Cortisol (via ACTH release) and epinephrine secretions arise from the adrenal cortex and medulla, respectively. Activation of the sympathetic nervous system increases both norepinephrine release (causing tremulousness, alertness and palpitations) and acetylcholine release (causing hunger and sweating). At the level of the pancreas, in response to hypoglycemia, endogenous insulin secretion from the β-cell is suppressed while glucagon release from the α-cell is elevated. By antagonizing insulin effects, these counterregulatory hormones work to increase blood glucose levels by stimulating endogenous glucose release from the liver, stimulating feeding behavior, and suppressing glucose clearance.
Hypoglycemia is an acute complication associated with the treatment of diabetes. Episodes of severe, temporarily disabling hypoglycemia, often with seizures or coma, occur yearly in 10-25% of people with Type 1 (insulin-dependent) diabetes.(1) As discussed below, there are many reasons why patients with both Type 1 and longstanding Type 2 diabetes have defective counterregulatory responses to hypoglycemia and are therefore susceptible to more frequent and more severe episodes of hypoglycemia. For these patients with diabetes, hypoglycemia becomes the rate-limiting step in the management of their blood sugar.(2) This barrier of hypoglycemia prevents people with diabetes from achieving the known microvascular benefits associated with intensive blood sugar control. This Cyberounds® will identify the key glucose sensing systems in the central nervous system and identify clinically proven therapeutic strategies to prevent hypoglycemia for patients with diabetes.
Sites of CNS Glucose Sensing
In order to detect slight fluctuations in glucose levels, the body is equipped with numerous sensors located throughout the body, including the gut, portal vein, pancreas and brain.(3)(4)(5)(6)(7) Many studies have confirmed that the brain serves as the body's primary glucose sensor and the hypothalamus functions as the hub of glucose sensing(8)(9)(10)(11). Within the hypothalamus, glucose sensing predominates in discrete regions, namely the ventromedial hypothalamus (VMH), arcuate nucleus (ARC) and paraventricular nucleus (PVN).
The VMH is the most studied region. Several studies abolishing VMH function have demonstrated its role in CNS glucose sensing. Specifically, creating lesions or chemical destruction of the VMH deregulated peripheral glucose homeostasis.(11)(12)(13) While all neurons metabolize glucose, a set of critically important neurons within the VMH and brain stem sense and respond to changes in blood sugar. By coupling their neuronal activity (i.e., firing rate) in response to their metabolism of glucose, these specialized neurons have been classified as "glucose sensing" neurons and not merely as "glucose using" neurons.
These glucose sensing neurons are categorized as glucose-excited neurons (GE), which increase their firing rate when extracellular glucose concentrations are elevated or glucose-inhibited neurons (GI), which are activated by decreases in extracellular glucose concentration or by cellular glucoprivation(14)(15)(16) (Figure 2).
Figure 2. Mechanism of Glucose Sensing in Glucose-excited (GE) Neurons.
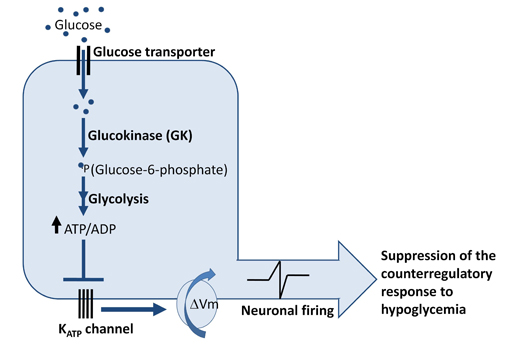
Glucose sensing in glucose excited (GE) requires glucose uptake via glucose transporters (GLUTs), glucose phosphorylation by the rate-limiting enzyme glucokinase, and subsequent metabolism of glucose to increase the intracellular ATP-to-ADP ratio. In the setting of glucose sufficiency, ATP-sensitive K+ channels are closed, the membrane is depolarized, and neuronal firing occurs, which suppresses the counterregulatory response while glucose levels are not low. Thus a rise in plasma glucose increases neuronal activity in GE neurons. However, when glucose levels fall as would occur during hypoglycemia, the decreased metabolism of glucose leads to opening of K-ATP channels and hyperpolarization of these glucose excited neurons. Conversely, another set of less well characterized glucose sensing neurons, glucose-inhibited (GI) neurons, are activated during hypoglycemia and are quiescent during hyperglycemia. ( ΔVm: change in membrane potential, membrane depolarization).
Both neuronal populations are widely distributed throughout the brain, but display distinct enrichment in hypothalamic and hindbrain regions classically associated with neuroendocrine regulation, energy homeostasis and nutrient metabolism.
Mechanism of Central Nervous System (CNS) Glucose Sensing
The mechanism of CNS glucose sensing is not clearly defined but appears to be dependent on the metabolism of glucose within key glucose sensing neurons. To better understand CNS glucosensing, researchers have drawn clues from more widely studied glucose sensing mechanisms, including the glucose sensing system that occurs in the pancreatic b-cell. Analogous to the pancreatic b-cell, GE neurons couple glucose metabolism to cell depolarization when extracellular glucose rises (Figure 2). Furthermore, the existences of b-cell glucose sensing proteins, including glucose transporters (GLUTs), glucokinase (GK), and ATP-sensitive K+ channels (KATP), have been reported in hypothalamic nuclei.(4)(16)(17) Therefore, it is speculated that these proteins may have a broadened role that includes CNS glucose sensing.
Glucose sensing neurons also play an important role in the detection of hypoglycemia as they are thought to be critical for the initiation of the counterregulatory response. As a consequence, these sensing proteins, among other hypothalamic nutrient sensors, may serve as therapeutic targets to combat hypoglycemia.
Facilitative Glucose Transporters (GLUTs)
Facilitative glucose transporters (GLUTs) support the passive influx of glucose into cells. Several GLUTs have been identified in the brain. (18)(19) Of these, GLUT1 and GLUT3 are the predominant isoforms expressed in the brain.(20) Although found in lower quantities, GLUT2 is expressed in key glucose sensing regions.(21)(22) Similar to its role in b-cell glucosensing, mounting evidence suggests that GLUT2 may be involved in hypothalamic glucosensing.(23)(24) GLUT4, the insulin-responsive glucose transporter, is expressed along with the insulin receptor in many important brain regions involved in glucose sensing,(25)(26) although its role as a brain glucose sensor remains unknown.
Glucokinase (GK)
The enzyme glucokinase (GK), a key regulator of glucose phosphorylation and metabolism in the b-cell, may play a role in the hypothalamus as a mediator of glucose sensing.(27) Glucokinase expression has been demonstrated in hypothalamic neurons by in-situ hybridization and RT-PCR.(28)(29) Further, modulating hypothalamic GK activity has been shown to regulate the counterregulatory response to hypoglycemia. Specifically, studies were performed to inhibit hypothalamic GK activity by injecting the glucokinase inhibitor alloxan or an adenovirus expressing a GK short hairpin RNA (to chronically reduce GK hypothalamic expression) into the ventricle adjacent to the hypothalamus. Results of these studies show that GK inhibition selectively enhanced the epinephrine response to hypoglycemia.(28) Conversely, enhancing GK activity with a glucokinase activator markedly suppressed the counterregulatory response.(28)
ATP-sensitive K+ (KATP) Channel
ATP-sensitive K+ channels (KATP) may provide a link between glucose metabolism and electrical activity in glucose sensing neurons. Several KATP channel subunits (Kir6.2, SUR1 and SUR2) have been localized in the hypothalamus.(30)(31) Also, the involvement of the KATP channel in central glucose sensing and counterregulation has been shown by several in vivo studies. Either intracerebroventricular (ICV) or direct VMH injection of the channel inhibitor glibenclamide blocks the counterregulatory response to acute hypoglycemia or anti-metabolite-induced glucopenia.(32) KATP channel inactivation (via Kir6.2 gene knockout) also leads to an impaired counterregulatory response to hypoglycemia. Further, KATP inactivity correlates to suppressed glucose-regulated firing activity of VMH neurons.(33) In contrast, activation of ATP-sensitive K+ channels in the VMH with channel opener diazoxide amplifies counterregulatory hormone responses to hypoglycemia in normal and recurrently hypoglycemic rats.(34)
AMPK
AMP-activated protein kinase (AMPK) is an important nutrient sensor present in the brain. When nutrient supplies are low, there is an increase in cellular AMP, which leads to allosteric activation of AMPK. AMPK activity stimulates catabolic pathways and suppresses anabolic pathways to restore energy homeostasis. In the hypothalamus, AMPK activity is increased in response to insulin-induced hypoglycemia and reduced by peripheral or ICV glucose infusion.(35)(36)
AMPK may also regulate the counterregulatory response to hypoglycemia. Enhancing VMH-AMPK activity with an activator, AICAR, markedly increases endogenous glucose production during a hypoglycemic clamp and improved the counterregulatory response in animals with defective counterregulation.(37)(38) Conversely, blocking hypothalamic AMPK with compound C, expressing a dominant negative form of AMPK, or reducing hypothalamic AMPK expression via RNA interference strongly reduced counterregulation to insulin-induced hypoglycemia.(39)
Additional Players: Neurotransmitters GABA & Glutamate
Two major classes of neurotransmitters in the brain, GABA (inhibitory) and glutamate (excitatory), have been postulated to play a role in CNS glucose sensing.(40)(41) Blocking GABA receptors in VMH stimulates a counterregulatory response to hypoglycemia,(41)(42) while blocking glutamate release in the VMH impairs the counterregulatory response to hypoglycemia.(40) These studies suggest that these neurotransmitters in VMH neurons provide an underappreciated role in CNS glucose sensing mechanism. In the future, efforts to prevent severe hypoglycemia might be achieved by modulation of these or other neurotransmitters in key brain regions to enhance the counterregulatory response.
Hypoglycemia and the Counterregulatory Response
Normal non-diabetic people have fasting blood glucoses <100 mg/dl and postprandial blood glucoses <135 mg/dl. The American Diabetes Association Working group on Hypoglycemia (43) defines hypoglycemia as a blood glucose ≤70 mg/dl (3.9 mmol/l). Whipple's triad provides a more relevant clinical definition of hypoglycemia: 1) the development of symptoms (autonomic or neuroglycopenic), 2) a low plasma glucose level, and 3) the reversal of symptoms when blood glucose is restored to a normal level.
In healthy young individuals, there is a hierarchical response to low blood sugar. Initially, when a person's blood sugar falls below 70 mg/dl, there is suppression of endogenous insulin secretion and the release of counterregulatory hormones (glucagon, epinephrine and growth hormone). If blood sugar drops <60 mg/dl, ACTH (and cortisol) are released provoking the development of autonomic symptoms which are mediated by the release of catecholamines (norepinephrine and epinephrine). Norepinephrine and epinephrine regulate symptoms of tremor, anxiety and palpitations. Acetylcholine release from sympathetic neurons results in additional symptoms, which include sweating, hunger and paresthesias (Figure 1). Finally, neuroglycopenic symptoms (impaired cognition, weakness, confusion, lethargy, coma or death) develop when blood glucose levels fall below 50 mg/dl.
The Diabetes Control and Complications Trial in 1993 (44) clearly demonstrated a reduction in the risk of long-term microvascular complications in Type 1 diabetics treated with intensive insulin therapy. The decrease in HgBA1C in these individuals, however, was associated with a two- to three-fold increase in the risk of hypoglycemia. Some of the risk factors for severe hypoglycemia include age (risk is greater in children as compared to adults), duration of diabetes, tight glycemic control, hypoglycemia unawareness, and previous episodes of severe hypoglycemia.(45)
Impaired Counterregulation and Hypoglycemia-associated Autonomic Failure (HAAF)
Type 1 and advanced Type 2 diabetes are characterized by insulin deficiency. Over time, insulin-dependent diabetic patients develop an impaired counterregulatory response to hypoglycemia. As compared to normal young healthy adults, in the setting of hypoglycemia, insulin-deficient diabetic patients do not have a decrease in endogenous insulin secretion that would normally occur in response to hypoglycemia. Secondly, following insulin deficiency, patients with diabetes lose the normal increase in glucagon secretion in response to hypoglycemia. The mechanism for this loss of glucagon response with insulin deficiency is unknown but may be due to impaired glucose sensing in the α-cell, a failure for a decrement in intra-islet insulin to augment glucagon secretion,(46) a failure for the decrement in intra-islet zinc ions, (i.e., perhaps not insulin) to augment glucagon secretion,(47) autonomic neuropathy unique to the pancreatic α-cell,(48), or chronic lack of insulin signaling on the α-cell.(49)
Without an increase in glucagon, insulin-deficient diabetic patients become exclusively dependent on the sympathoadrenal response to hypoglycemia. Eventually, insulin-deficient diabetic patients also develop an attenuated sympathoadrenal response to hypoglycemia which leads to hypoglycemia unawareness.(50) This puts patients at a 25-fold or greater risk for severe iatrogenic hypoglycemia.(51)
Hypoglycemia-associated autonomic failure (HAAF) is characterized by a decrease in the epinephrine response to a given level of hypoglycemia caused by recent antecedent iatrogenic hypoglycemia.(51) Its two components include a defect in glucose counterregulation (i.e., attenuated sympathoadrenal response to hypoglycemia in the setting of absent increases in glucagon and decreases in insulin) and hypoglycemia unawareness (lack of symptoms to a given level of hypoglycemia). This is a vicious cycle that leads to recurrent episodes of severe hypoglycemia that often go unnoticed by patients.(51)
Exercise and Sleep-associated Hypoglycemia Unawareness
In addition to antecedent iatrogenic hypoglycemia, antecedent exercise and sleep have been shown to lead to HAAF.(52) Antecedent exercise causes a reduction in the epinephrine response to hypoglycemia. This leads to an impaired counterregulatory response which has not been shown to be associated with a decrease in hypoglycemia awareness.(49) During sleep, it has also been demonstrated that Type 1 diabetic patients have a decreased sympathoadrenal response to hypoglycemia and that they are less likely to be awakened during sleep by hypoglycemia than are non-diabetic patients.(49)
Treatment and Prevention of Hypoglycemia in Diabetic Patients
According to the American Diabetes Association (ADA), 15-20 g of glucose is the best treatment for hypoglycemia in an individual who is awake and conscious.(53) After initial treatment a patient should recheck his or her blood glucose in 15 minutes and repeat treatment if needed. Once an individual's blood sugar returns to normal, he or she should eat a meal to prevent further episodes of hypoglycemia. All patients at risk for hypoglycemia should be prescribed a glucagon emergency kit and caretakers should be instructed on its use. In addition, the ADA recommends that patients with hypoglycemia unawareness avoid further episodes for several weeks by increasing their glycemic targets. This treatment has been shown to reverse hypoglycemia unawareness and reduce the risk of further episodes of severe hypoglycemia.(50)
Clinicians must also be aware of and educate their patients about ways to avoid hypoglycemia. It is well known that subcutaneous insulin is not physiologic and therefore has many pitfalls. Although insulin analogs have been designed to control fasting and post-prandial hyperglycemia, they all have the potential to cause iatrogenic hypoglycemia. The short-acting insulin analogs (insulin aspart, insulin lispro and apidra) have been shown to control post-prandial blood glucose more effectively than regular insulin, and because of their shorter biological half-life they are less likely to cause post-prandial hypoglycemia. A large meta-analysis (54) of greater than 1400 patient-years showed a 25% reduction in the frequency of severe hypoglycemia with insulin lispro compared with regular insulin. In a randomized control trial (55) where 56 Type 1 diabetic patients were treated with either lispro or regular insulin as their mealtime insulin plus NPH for one year, HgBA1C was lower in the group treated with insulin lispro (6.34% vs. 6.71%, p <0.002), and there were fewer episodes of hypoglycemia (7.4 vs. 11.5 episodes/patient-month).
In addition to choosing a short-acting mealtime insulin, patients and clinicians must select a long-acting basal insulin treatment. When NPH is given before the evening meal (alone or in combination with short-acting insulin, as in 70/30 mixed insulin), it peaks in the middle of the night and can increase the risk of nocturnal hypoglycemia. In a randomized control trial (56) of 534 reasonably well-controlled Type 1 diabetic patients (mean HgBA1C 7.7%) who received pre-meal regular insulin, and either insulin glargine at bedtime or NPH insulin once or twice a day for one month, fewer patients in the glargine group experienced symptomatic hypoglycemia (39.9% vs. 49.2%, p=0.0219) and nocturnal hypoglycemia (18.2% vs. 27.1%, p=0.0116) after one month of treatment. Thus, for insulin-treated diabetic patients, the newer insulin analogues that combine long-acting basal insulin coverage with a rapid acting pre-meal insulin regimen may decrease the overall risk of hypoglycemia.
Use of Continuous Subcutaneous Insulin Infusion (CSII) vs. Multiple Daily injections (MDI)
Continuous subcutaneous insulin infusion via an insulin pump is rapidly gaining popularity for the treatment of many patients with diabetes. An insulin pump eliminates the need to administer multiple daily injections and theoretically would be associated with fewer episodes of hypoglycemia than multiple daily injections. Studies have not, however, conclusively demonstrated that CSII decreases the risk of hypoglycemia.
There is research (57) that shows that CSII is associated with a decreased risk of hypoglycemia in adults but not in children. One such study compared NPH to CSII (58). In this study, 272 patients were randomized to treatment with CSII or MDI with NPH as the basal insulin. After six months of treatment, there was a reduction in the incidence of mild hypoglycemia in the CSII group as compared to the MDI group (mean frequency with MDI 55.4 events per patient years vs. 49.3 events per patient years, p=0.001), as well as a reduction in the incidence of severe hypoglycemia, usually defined as hypoglycemia requiring assistance from another individual (0.5 events per patient year with MDI vs. 0.2 events per patient year with CSII, p < 0.001).
As compared to MDI with insulin glargine, on the other hand, CSII has not been demonstrated to be associated with a decreased risk of hypoglycemia. In a randomized control trial (59) of 107 adults with insulin-treated T2DM and a mean HgBA1C of 8.2% treated with either CSII or MDI with lispro and glargine for 12 months, there was no difference in the rates of hypoglycemia or in the reductions in HgBA1C. Hirsch et al. did not find a difference in the percentage of hypoglycemic or nocturnal hypoglycemic episodes in their study of 100 patients randomized to either MDI with aspart and glargine or CSII.(60) In contrast to these randomized-control studies, a meta-analysis (61) comparing the rates of severe hypoglycemia with MDI vs. CSII showed a reduction in rates of severe hypoglycemia in patients using CSII. This reduction was more profound the longer the duration of diabetes.
Continuous Glucose Monitoring (CGM)
Continuous glucose monitoring allows for patients to receive continuous, virtually real-time, feedback on their blood sugar trends and detects hypo- and hyperglycemia that a patient may not otherwise be aware of (i.e., at night). A continuous glucose monitoring system is a portable device that measures glucose in the interstitial fluid and provides measurements of glucose every one to five minutes (up to 288 measurements of glucose per day). Most monitors have to be calibrated at least four times per day with measurements of capillary blood glucose.
Although studies have shown an improvement in HgBA1C with the use of continuous glucose monitors, most have been of short duration and have not had an adequate control group. No study has looked at the incidence of hypoglycemia as a primary endpoint. A meta-analysis recently published(62) showed a non-significant reduction in HgBA1C with CGMS compared to SBGM (self-blood finger-stick glucose monitoring). Because of variability in defining hypoglycemia, the authors were unable to compare the rates of hypoglycemia in both groups. They did find, however, a trend towards decreased nocturnal hypoglycemia with CGMS. Finally, in a multicenter clinical trial (63) in which 322 well controlled adults and children with Type 1 DM (mean HgBA1C 7.6-8.0%) were randomly assigned to use continuous glucose monitoring or monitoring with a blood glucose meter, there was an improvement in HgBAIC only in patients ≤25 years of age and no difference in the rates of hypoglycemia. One must keep in mind, however, that this study was not powered to detect a difference in the rate of hypoglycemia and the rates of hypoglycemia were low.
New Directions
New treatments on the horizon for hypoglycemia include pancreas and islet cell transplantation and pharmacologic treatments aimed at increasing the counterregulatory response to hypoglycemia. Although associated with significant morbidity and some mortality, whole pancreas transplantation may be a treatment option for patients who suffer from severe iatrogenic hypoglycemia.(50) Reported symptoms of hypoglycemia and documented episodes of severe hypoglycemia are rare after pancreas transplantation. In response to hypoglycemia, glucagon secretion returns to normal, and epinephrine and growth hormone responses improve but do not return to normal.(64)
Islet transplantation, a less invasive procedure, also results in insulin independence and fewer episodes of severe hypoglycemia. According to a survey of all North American transplant programs by the collaborative islet transplant registry, less than 5% of transplant recipients had one or more episode of severe hypoglycemia in the first year following transplant as compared to greater than 85% prior to transplant.(65)
Finally, several agents are currently being investigated as treatments to prevent hypoglycemia by increasing the counterregulatory response. Preliminary, small studies have shown some degree of efficacy for agents such as caffeine, theophylline, modafanil and SSRI's, (66) although these agents have not been assessed in large randomized trials.
Conclusion
Hypoglycemia continues to be a barrier in achieving glycemia control for patients with Type 1 and advanced Type 2 diabetes. Impaired brain glucose sensing limits the appropriate sympathoadrenal counterregulatory response and contributes to hypoglycemia unawareness, thus increasing the frequency and severity of hypoglycemia for patients with diabetes. Progress in the basic science unraveling the molecular biology of neuronal glucosensing has already identified potential key targets that may be amenable to pharmacologic intervention to augment the counterregulatory response to hypoglycemia in diabetic patients.
Clinical trials are needed to determine if an enhancement of the counterregulatory response may allow patients with diabetes to achieve the benefits associated with intensive glycemic control, while minimizing the potentially catastrophic consequences of hypoglycemia. Fortunately, many advances in glucose monitoring and progress to more physiological insulin delivery have been shown to reduce this barrier of hypoglycemia, and thus enhancing the quality of life for people with diabetes.