Course Authors
Frieda Wolf, M.D.
Release Date: 11/21/2008
Upon completion of this Cyberounds®, you should be able to:
Describe the zipper-like structure and some components of the glomerular basement membrane
Discuss congenital forms of nephrotic syndrome
Discuss the clinical-pathologic classification of various nephrotic syndromes
Discuss various therapies for nephrotic syndrome.
'bubbles appearing on the surface of the urine indicate renal disease and a prolonged illness discloses chronic inflammatory activity in the glomeruli and may be identified with the term chronic glomerulonephritis'
Hippocrates, Aphorisms VII, 34(6)(1)
Although nephrotic syndrome was described by Hippocrates, it was not until 1764 that Domenico Cotugno made the observation that 'a dense white body like albumen of boiled eggs was found in the urine after heating.'Almost 30 years later (1790), Cotugno published that the urine of some subjects with dropsy (the former term for edema) coagulated on heating.(2) In 1827 Richard Bright related 'dropsical urine' to kidney disease, 'I have never yet examined the body of a patient dying of dropsy attended with coagulable urine in whom some obvious derangement was not discovered in the kidneys.'(3)
Dropsy was the term used to describe anasarca, regardless of cause, during the 16th to the 18th centuries. The term 'nephrosis' was introduced by Muller, a German pathologist, in an attempt to differentiate nephritic kidneys -- which were inflamed and exudative -- from non-inflammatory diseases, which were parenchymal, and made the kidneys appear fatty. This was incorporated into the classification of Bright's disease in 1914 by Fahr and Volhard. Nephrosis, however, was still generally believed to be a tubular disorder at that time. The term nephrotic syndrome was first used in the 1920s and became routine only after the advent of renal biopsies in the mid 1950s.(4)
What is Nephrotic Syndrome?
Today, nephrotic syndrome is defined as a combination of albuminuria with hypoalbuminemia, leading to edema, accompanied by hyperlipidemia and lipiduria. Approximately 3.5 grams of albuminuria in 24 hours define nephrotic range proteinuria. The pathophysiology of nephrotic syndrome has recently been elucidated though improved understanding of the glomerular basement membrane (GBM) structure and function. Furthermore, variable clinical outcomes have been explained by mutations of different molecules in the GBM.
The Glomerular Basement Membrane
The GBM is composed of three layers: the fenestrated endothelial cells lining the inside of the glomerular capillary; the glomerular basement membrane (GBM) and the podocyte; and foot processes with their intervening slit diaphragm.(5) The slit diaphragm constitutes a porous filter structure of nephrin molecules from two adjacent foot processes. Nephrin is a transmembrane protein with a large extracellular domain-like immunogobulin. Nephrin molecules extend toward each other from neighboring foot processes and dimerize across the slit diaphragm.(6) Nephrin connects to podocin (another protein connecting nephrin to the foot process in the slit diaphragm) and ultimately the actin cytoskeleton, within the cytoplasm of the foot processes. These proteins help maintain glomerular permeability.(7) CD2-Associated Protein (CD2AP) interacts with nephrin and possibly also with podocin. It also links the actin cytoskeleton between two filtrations slits with other proteins.
The GBM is a highly crosslinked macromolecular meshwork composed of extra-cellular matrix proteins such as type IV collagen, laminins, nidogen and heparan sulfate proteoglycans. Microtubules and intermediate filaments make up its cytoskeleton. A number of proteins attached to actin filaments (e.g., talin, paxillin and vinculin) mediate the link between cytoskeleton and GBM integrins. The GBM is flanked by endothelial cells on one side, and by podocytes, which are visceral epithelial cells, on the other.(8)

Figure 1. Cartoon of the Glomerular Basement Membrane.
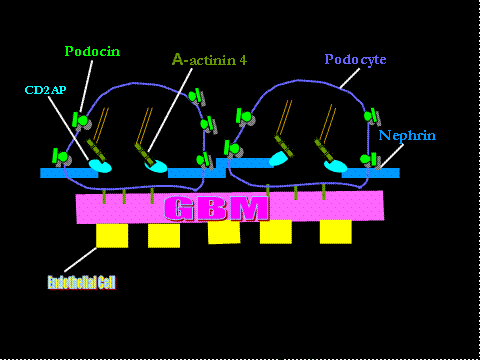
Courtesy of the author.
Genetic Mutations and Their Consequences
Alpha-actinin-4 is a crosslinking protein that interacts with intra- and intercellular adhesion molecules and elements of the transmembrane signal transduction pathway. It stablilizes the actin cytoskeleton by crosslinking actin filaments (F-actin). It is interesting to note that mutations in nephrin, CD2AP or podocin lead to proteinuria and nephrotic syndrome, whereas mutations in alpha-actinin-4 result in focal segmental glomerular sclerosis.(9)
Rare cases of inherited defects, nephrin deficiency being the most common, can lead to nephrotic syndrome. Nephrin is encoded by the NPHS1 gene located on chromosome 19. NPHS1 mutations result in the congenital nephrotic syndrome of the Finnish type (CNF), which was first described in 1956. CNF is a rare autosomal recessive disease, occurring predominantly in Finland, with an incidence of 1 per 8,200-10,000 live births. Proteinuria presents in the neonatal life, along with dysfunction of podocyte foot processes, irregular distention of renal proximal tubes and large placenta, with its weight usually exceeding the neonate's total body weight. CNF is resistant to treatment with steroids or alkylating agents. Survival of neonates is dependent on dialysis with a subsequent kidney transplant. Proteinuria recurs in 25% (most likely due to the high titer of anti-nephrin antibodies).
The NPHS2 gene encodes podocin in the slit diaphragm and dysfunction of this protein will also lead to proteinuria. Wilms' tumor suppressor gene (WT1) is expressed in podocytes and is required for podocyte function. WT1 mutations are seen in sporadic Wilms' tumors and in association with Frasier and Denys-Drash syndromes. Both syndromes are characterized by gonadal dysfunction and progressive nephropathy with FSGS, or diffuse mesangial sclerosis with onset in early childhood.(10) Mutations of the mitochondrial genome can also cause podocyte malfunction and lead to FSGS.
Another organelle, the lysosome, may also be responsible for glomerular disease. Fabry disease is caused by mutations of the GALA gene encoding galactosidase A. The galactosidase A deficiency results in globotriaosyloceramide deposition in lysosomes of all types of the glomerular cells, especially in podocytes and epithelial cells of distal tubular. Clinically, there is proteinuria and progression to end-stage renal disease. Fabry disease may be specifically treated with an enzyme-replacement therapy.
With the improved understanding of the structure and function of the GBM and the identification of specific molecules which are damaged in nephrosis, specific clinical outcomes can now be explained. For example, steroid-resistant FSGS largely results from genetic mutations of the GBM. Podocyturia is common to various nephrotic syndromes including those secondary to systemic disease such as diabetes. Podocyte damage is a central precondition of proteinuria, as is nephrin deficiency or mutation. This molecular clarification helps to elucidate the clinical division of nephrotic syndromes, which traditionally have been classified by pathologic appearance.
Classification of Nephrotic Syndrome
Nephrotic syndrome may be idiopathic or secondary to a known disease (secondary nephrotic syndrome). The most common known causes of nephrotic syndrome are diabetes mellitus, amyloidosis, systemic lupus erythematosus, drugs and malignancies. The various nephrotic syndromes have common clinical presentations, such as edema, hyperlipidemia, thrombosis and predisposition to infection.
Nephrotic syndrome can also be classified into five types according to pathological findings. The original intention of this classification system was to sort idiopathic causes according to pathology types, but many types previously thought to be idiopathic now have known causes:
- Minimal change disease (lipoid nephrosis)
- Focal segmental glomerulosclerosis
- Membranous nephropathy
- Membranoproliferative glomerulonephritis
- Proliferative glomerulonephritis (mesangial, IgA nephropathy).
We will not discuss diffuse proliferative glomerulonephritis and mesangial proliferative GN because they are less often associated with nephrotic syndrome. However, some proliferative GN may be accompanied by nephrotic syndrome. For example, about 20% of patients with post-streptococcal glomerulonephritis (which typically presents as diffuse proliferative GN) develop a nephrotic range of proteinuria. Similarly, mesangial proliferative GN may be accompanied by massive proteinuria. Even Alport's disease, which manifests mainly as interstitial nephritis, may have a nephrotic range of proteinuria.
Nephrotic Syndrome: Clinical Manifestations
The clinical manifestations of nephrotic syndrome include proteinuria, which in itself may be toxic to tubular cells and the interstitium, edema, thrombotic tendency leading to deep vein thrombosis (DVT) or renal vein thrombosis. Infections and acute renal failure are also common.
Proteinuria
Severe proteinuria is a requirement for nephrotic syndrome and it is arbitrarily defined as more than 3.5 g/24 hours. However, the degree of proteinuria that causes the clinical manifestations of nephrotic syndrome varies with the nutritional state of a patient. A well nourished patient losing 5 g of protein per day may not even develop hypoalbuminemia, while a malnourished person losing only 3 g per day may have severe hypoalbuminemia with all the manifestations of nephrotic syndrome.
The magnitude of proteinuria is documented by collecting a 24-hour urine sample. However, because creatinine excretion rate tends to be fairly constant, a simultaneous measurement of urine creatinin.
Proteinuria is classified into three types:
Glomerular proteinuria is caused by increased filtration of proteins at the glomerulus, and this is the type seen in nephrotic syndrome and most renal diseases.
Tubular proteinuria is caused by decreased reabsorption of normally filtered protein (such as beta-2-microglobulins and retinol-binding protein) by the proximal tubules. This may occur with tubulointerstitial diseases that affect the proximal tubular function.
Overflow proteinuria occurs when the amount of filtered proteins exceeds the normal capacity of the tubules to reabsorb them. An example is light chain excretion in multiple myeloma. It can also occur when a large amount of albumin is rapidly infused.
Selective or Non-selective Proteinuria
In selective proteinuria, albumin is primarily excreted; in non-selective proteinuria, proteins of different molecular sizes are indiscriminately excreted. Selectivity of proteinuria is determined by measuring relative clearance of gamma globulin (representing proteins of large molecular weights) and albumin (representing proteins of small molecular weights). A clearance ratio less than 0.1 represents selective proteinuria, while a ratio greater than 0.5 indicates non-selective proteinuria. Selective proteinuria tends to suggest minimal change disease and non-selective proteinuria suggests other pathological types. However, there is much overlap.
Edema
Edema of nephrotic syndrome (nephrotic edema) is caused by a reduction in the oncotic pressure of the plasma, which provokes an extracapillary shift of water. This shift leads to secondary renal retention of salt and water and, thereby, extracellular volume expansion. Filtration capacity of capillaries is doubled in nephrotic patients.
Another mechanism of nephrotic edema is primary renal salt retention in the cortical collecting duct (CCD) from the renal disease itself. Activation of Na/K ATPase on the basolateral membrane of principal cells in the CCD actively extrudes Na and creates a gradient so that epithelial sodium channels (eNacs) on the opposite side can passively drive Na. (This is the primary mechanism for the salt retention seen in nephrotic syndrome in contrast to the salt retention of heart failure, which is caused by aldosterone activation.)
Role of Hyperlipidemia
Hyperlipedima maintains oncotic pressure in the face of the protein loss characteristic of the nephrotic syndrome. In nephrotic syndrome, LDL cholesterol and triglycerides are high and HDL cholesterol is normal or low. High LDL cholesterol occurs as a result of down regulation of LDL receptors and up regulation of ACAT (acyl-CoA cholesterol acyl transferase) in the liver. These two effects reduce hepatic free cholesterol concentration and increase cholesterol synthesis by up-regulating HMG-CoA reductase, the rate limiting enzyme for cholesterol synthesis. Lethicin cholesterol acyltransferase (LCAT) is responsible for the extrahepatic uptake of cholesterol by cholesterol-poor HDL-3, which is then converted to cholesterol-rich HDL-2 and results in low serum HDL. Deficiency of LCAT, caused by its urinary loss in association with proteinuria, occurs in the nephrotic syndrome. Finally, VLDL is high because of impaired clearance of VLDL.(11) The overall result is increased synthesis of lipoproteins, abnormal transport of circulating lipids, increase in lipoproteins associated with LDL, and decrease in those associated with HDL.
Thrombotic Tendency and Infection
Renal vein thrombosis, as well as thrombosis of systemic veins, is a common finding in nephrotic syndrome. Excessive production of coagulation factors such as factor VIII and fibrinogen, loss of anti-coagulant factors such as antithrombin III and protein S in the urine, as well as antigen-antibody complexes are thought to be responsible for the increased incidence of thrombosis. Renal vein thrombosis is common in all types of nephrotic syndrome but is particularly frequent in membranous nephropathy and membrano-proliferative nephropathy, probably because immune deposits in the GBM promote clotting.
Impaired general nutrition attributed to severe protein loss is also likely to contribute to the increased incidence of infection.
Prognosis and Therapy
Nephrotic syndrome occurs either as a renal manifestation of a systemic disease such as diabetes, lupus or amyloidosis, or as a primary renal disease. Membranous nephropathy (MN) and focal segmental glomerulosclerosis (FSGS) are the two most common pathological types of nephrotic syndrome among adults; Minimal Change Disease is the most common in children.
Though the prognosis and therapy for nephrotic syndrome types differ (see below), some common therapeutic principles for proteinuric disease exist. The use of Angiotensin-Converting Enzyme (ACE) inhibitors in combination with Angiotensin Receptor Blockers (ARBs) for proteinuria and hypertension is paramount. Diuretics for edema and lipid lowering agents for the extremely elevated lipids of nephrotic syndrome are universal, except perhaps in minimal change disease. The use of dialytic support in cases with concomitant acute renal failure may also be appropriate. Treatment of secondary nephrotic syndrome employs these strategies, as well as specific treatment of underling disease. For example, in diabetic nephropathy, control of glycemia and hypertension are primary. In lupus nephritis, specific immune-modulating therapy is indicated.
The following discussion is based on the pathologic classification of nephrotic syndromes that was advocated after renal biopsy came into vogue in the 1950s. This classification proved useful for therapeutic purposes.
Minimal Change Disease (AKA Lipoid Nephrosis)
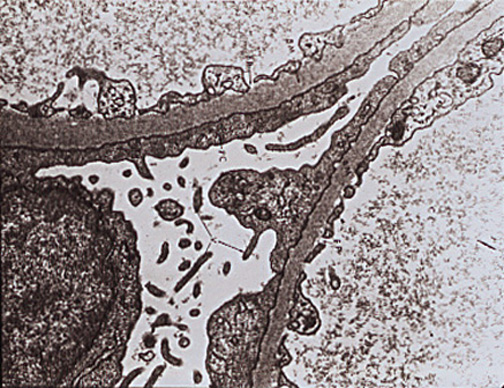
As mentioned, Minimal Change Disease (MCD) is, by far, the most common pathologic type of nephrotic syndrome in children. Its cause is unknown. Light microscopic examination is normal (Figure 3), and immunofluorescence is negative. Electron microscopy shows fusion of foot processes (Figure 4). A congenital variant, produced by a nephrin mutation, is characterized by a highly selective, heavy proteinuria. A defect in the visceral epithelial cell (podocyte) is hypothesized(12) but the prognosis is good.
The treatment of MCD remains glucocorticoids, as it is the only nephrotic syndrome truly responsive to steroids. It is, in fact, divided clinically into steroid-responsive and steroid-non-responsive types, the latter having worse prognosis because it often recurs or progresses to FSGS. Glucocorticoids are initially used in children and adults, although the prognosis in adults is inferior. No course of treatment has been identified as superior, though prednisone seems to confer more sustained remissions as compared with a short course of IV methylprednisolone.(13) In some adults, cyclosporine A is used, even in steroid-responsive MCD, as it seems to induce long-term remission.
Figure 2. Child With Periorbital Edema From Minimal Change Disease.
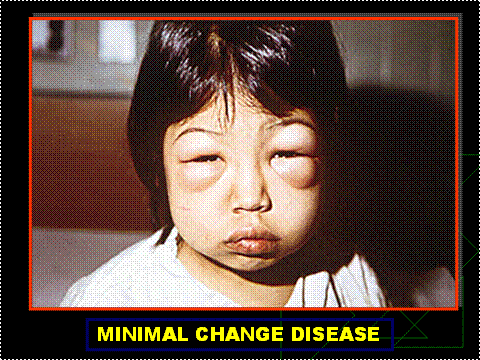
Figure 2. Minimal Change Disease: Normal Glomerulus Under Light Microscopy.
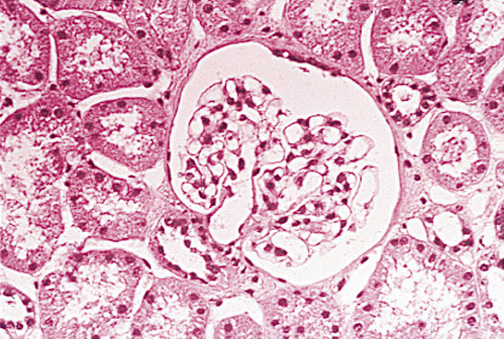
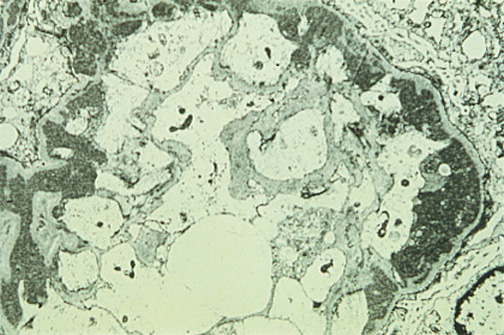
Figure 4. Minimal Change Disease: Electron Microscopy Shows Effacement and Sloughing of Foot Processes.
Focal Segmental Glomerulosclerosis (FSGS)
FSGS is mostly idiopathic but can be superimposed on other renal disease, e.g., IgA nephropathy. It can be associated with reflux disease or unilateral renal agenesis, can be secondary to HIV, heroin, obesity and even inherited in the familial variant. The incidence of FSGS is particularly high among black people.
Sclerosis of some glomeruli (focal) and parts of glomeruli (segmental) occurs. No consistent pattern of immune complex deposits and no inflammatory cell proliferation are seen. Occasionally, deposits of IgM and complements are present. The suspected pathogenetic mechanism is injury to podocytes. Visceral epithelial cell (AKA podocyte) damage is the hallmark.
Therapeutic response to steroids is poor and ~50% of FSGS patients progress to end-stage renal disease (ESRD). FSGS may also be a progressive lesion of MCD, especially if associated with deposits of IgM and C3 and injury to visceral epithelium.
A circulating factor is suspected -- the best characterized candidate in these steroid-sensitive patients is haemopexin, a100 kDa haem-binding protein.
Figure 5. Early FSGS With Partial Sclerosis in Glomeruli, While Tubules and Interstitium Remain Normal.
Compared to MCD, there are more treatment options for FSGS, (14)(15)although prognosis is inferior. Cyclosporine is the mainstay of immunotherapy and is more effective than glucocorticoids. Minimal treatment is 12 months and renal biopsy to monitor for nephrotoxicity may be useful, along with minimization of serum levels. Tacrolimus has also been tried successfully; however, no randomized clinical trials have been published to compare it with cyclosporine A or glucocorticoids.(16)
Mycophenolate mofetil has also been used in patients unable to tolerate cyclosporine,(17) but no randomized trials have been reported. Recurrence of FSGS in transplanted kidneys occurs in approximately 25% of individuals. Because of the hypothesized circulating factor in FSGS, plasmapheresis should be attempted for purported 'removal' of the offending cytokine, though its success has been variable.(18)
Membranous GN (Membranous Nephropathy)
The hallmark of the disease is diffusely thickened basement membrane. On light microscopy, the glomerular capillary wall is thick and on EM deposits are visualized on the subepithelial side of the basement membrane. These deposits appear granular with immunofluorescence (IF). With time, new basement membrane grows between deposits, eventually incorporating them into the basement membrane. Diseases known to cause membranous GN include lupus, chronic hepatitis B infection and various cancers. Exposure to toxins, such as penicillamine and mercury have also been implicated.
The pathogenetic mechanism of proteinuria is epithelial cell injury caused by activated complements. Autoantibodies to megalin complex (receptor complex that reabsorbs proteins) are suspected.(19) Approximately 40% of MN patients progress to renal failure.
Figure 6. Cartoon of Membranous Nephropathy.
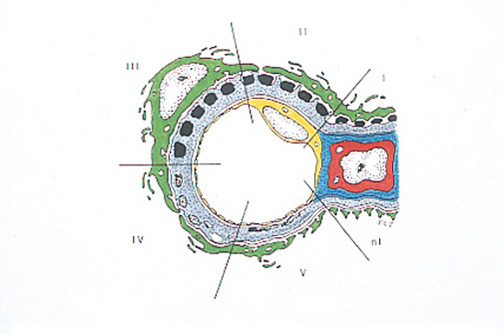
In Stage I, there are subepithelial, electron dense deposits with no projection of basement membrane; Stage II: well-defined projections of basment membrane between deposits; Stage III: deposits are surrounded by basement membrane; Stage IV: electron dense material fades creating 'holes' in the GBM; Stage V: repair of membrane occurs.
Figure 7. Patient With Membranous Nephropathy.
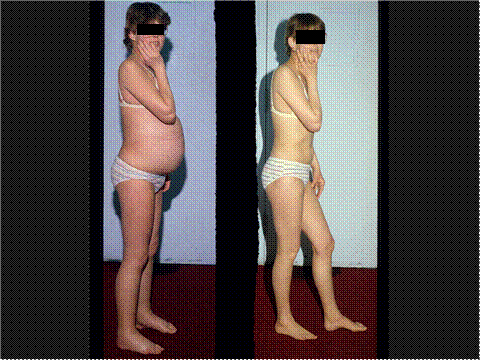
In relapse on left, in remission on right.
Similar to FSGS, treatment of MN(20) includes use of glucocortioids, cyclosporine A, mycophenlate mofetil and tacrolimus. In secondary membranous nephropathy, treatment of underlying disease is indicated, for example, use of mycophenolate in lupus nephritis.
MN recurs in approximately 40% of transplanted individuals.(21) Though MN seems to respond to additional immunosuppressive therapy, it is, nevertheless, progressive.
Membranoproliferative GN (MPGN)
Because light microscopy shows thickening of the basement membrane. as well as proliferation of mesangial cells, this type of nephrotic syndrome is named membranoproliferative GN. The membrane thickening is caused by interposition of mesangial matrix in a tram track appearance.
There are two types of MPGN: type I and type II. In type I, immune complexes and complements are deposited subendothelially in the GBM. In type II, electron dense deposits of unknown nature (speculation is that it is a lipid) are found in the basement membrane. Complement component 3 (C3) is also seen in the mesangium and basement membrane, but no IgG or earlier complement component, and this suggests that the classical complement pathway is not activated. Type II MPGN is also called dense deposit disease.
In type I MPGN, early and late complement components are often low. In type II MPGN, C3 is low but the early components of complements are normal. The mechanism of low C3 is activation of C3 through an alternative pathway, an autoantibody called C3 nephritic factor. Normally, a small amount of C3 is broken down to C3a and C3b without the help of immune complexes; C3b then combines, thanks to factor D, with factor Bb, forming C3bBb. This is C3 convertase, which can create more C3b. C3bBb is then quickly broken down with help of factor H. When C3 nephritic factor, the autoantibody to C3bBb, binds C3bBb, this enzyme is stabilized, and cannot be attacked by factor H. Hence, the alternate pathway is perpetually activated, resulting in consumption of the C3 and thus MPGN. Congenital deficiency of factor H also produces MPGN.
Figure 8. Alternate C3 Pathway.

Adipose tissue can be affected by perpetual activation of complements, and many patients with type II MPGN also develop partial lipodystrophy (usually manifested as loss of fat in the upper body and trunk). Causes of MPGN are idiopathic or secondary to known diseases such as chronic hepatitis C or B infection, and systemic lupus.
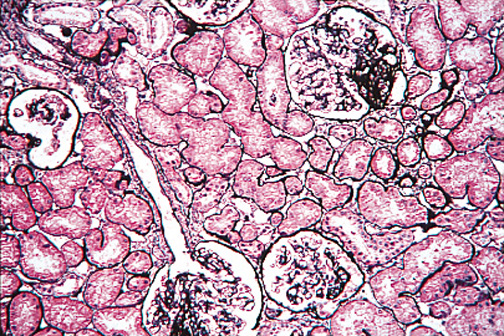
Figure 9. Type I MPGN.
Subendothelial deposits of immune complexes in type I MPGN
Figure 10. Type II MPGN.
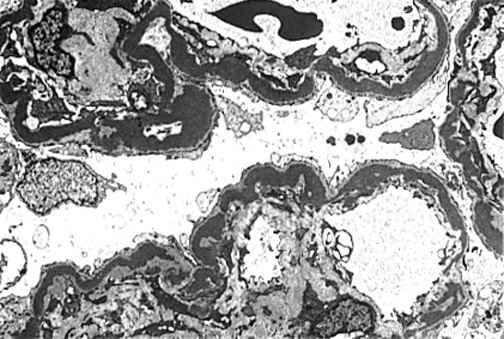
Note ribbon-like dense deposits in GBM.
Figure 11. Diffuse Proliferative GN Under Light-microscopy.
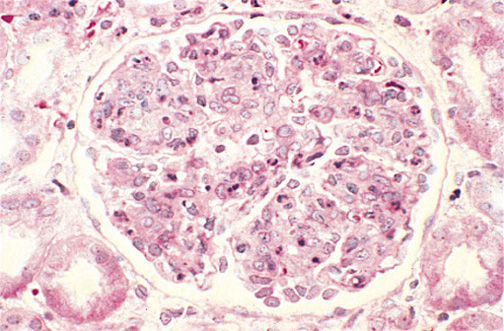
Glomerulus appears 'swollen' with matrix and cells. Polymorphonuclear cells indicate severe inflammation.
Figure 12. Tram Tracks of GBM.
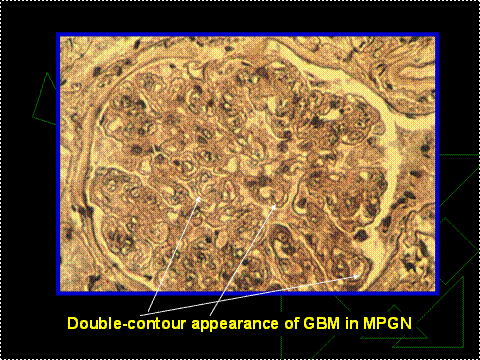
Note the double contour (white arrows) which resembles tram tracks.
Prognosis is pretty poor in MPGN. Even with treatment, 60% of patients progress to ESRD. Recurrence in transplanted kidney is almost universal with proteinuria and progressive lesions consistent with MPGN. As with native kidneys, however, only about 40% suffer graft loss.
Treatment of MPGN depends on the underlying cause. Treatment of hepatitis C can induce remission (22) and should be undertaken, if it is the cause, prior to kidney transplantation. In idiopathic MPGN, immunosuppressive therapy is employed; (23) cyclosporine A and tacrolimus have also been used. Since MPGN is often a disease in children, long-term use of steroids is not advisable and, instead, mycophenolate and cyclosporine are prescribed.(24)
Summary
Nephrotic syndrome has been documented in human history for millennia. As a result of insightful observations on the presence of albumin in urine, the use of renal biopsy and advances in immunology and renal physiology, we now have available some successful therapies.
Though the pathologic types of nephrotic syndrome, minimal change disease, focal segmental glomerulosclerosis, membranous glomerulopathy and membranoproliferative glomerulonephritis, have distinct clinical and therapeutic distinctions, they also have some common features -- podocyte dysfunction or activation of the immune system. The use of modern pharmaceutical therapy (e.g. ACE inhibitors and angiotensin receptor blockers) has been shown to retard progression of chronic kidney disease as a consequence of their anti-proteinuric effects. Immune-modulating therapies such as glucocorticoids and cyclosporine A have enabled long-term remission of nephrosis.
With advances in genetics, we can now identify subsets of patients whose prognosis is worse and tailor therapy for those individuals at high risk for recurrence and progression to ESRD. And, finally, as a result of our growing molecular biological knowledge of the glomerular basement membrane apparatus, we can now describe targeted proteins which have potential implications for therapeutic interventions.