Course Authors
Edward J. Bertaccini, M.D.
Release Date: 01/15/2007
Upon completion of this Cyberounds®, you should be able to:
Describe a brief history of the study of anesthetic mechanisms, from lipid theories to more modern protein theories of anesthetic action
Summarize the Meyer-Overton correlation and hypothesis, the exceptions to the Meyer-Overton correlation, the evidence for direct interactions of anesthetics with proteins
Discuss the amphipathic nature of the anesthetic binding site identified within several protein complexes
Discuss the theoretical construct of modern models of ligand-gated ion channels and their relationship to an anesthetic site of action.
For over 160 years, general anesthesia has been provided for the safety and comfort of a myriad of otherwise painful procedures, most notably surgery. Early investigations into the mechanisms of anesthetic action by Meyer and Overton(1) demonstrated a significant correlation of an anesthetic's potency with its lipid solubility in olive oil. The inference from such studies was that anesthetics must act by altering the properties of a larger source of lipid in the body, the cell membrane. This became known as the Meyer-Overton Hypothesis.
Since then, many studies have been performed to further characterize the more specific lipid environment in which anesthetics could act. These have included studies of anesthetic solubility in a variety of solvents.(2),(3) Other studies have sought to elucidate the exact effect of an anesthetic on the characteristics of membrane bilayers. Some have shown significant effects of anesthetics on parameters such as membrane expansion,(4) lipid order(5) and bilayer membrane lateral pressure profile.(6) While clearly showing an anesthetic effect, the majority of these studies have, unfortunately, been carried out at anesthetic concentrations far beyond those that are clinically relevant.
Exceptions to the Meyer-Overton Correlation
Until recently, the Meyer-Overton correlation of anesthetic potency with lipid solubility has remained a robust physical phenomenon. While the correlation is yet to be completely explained and accounted for, the original hypothesis stemming from that correlation now has some gross exceptions. Koblin and Eger(7),(8) have clearly demonstrated several classes of compounds that deviate from the Meyer-Overton rule. These include a multitude of polyhalogenated linear and cyclic hydrocarbons, whose lipid solubilities would predict reasonable anesthetic potency but whose actual effect is slim to none. In particular, with regard to the minimum alveolar concentration (MAC) of anesthetic gas required to suppress motion to surgical incision in 50% of the patient population, these substances can be classified into those with partial anesthetic effect (the transitional compounds) and those with no effect at all (the nonimmobilizers). In fact, within the series of transitional anesthetic compounds, potency correlates with a compound's hydrophilicity and not its lipophilicity. Other exceptions to this correlation include the differing potencies of various chemical isomers despite similar lipophilic characteristics.
Since the advent of the anesthetic phenomenon, there have also been vast advances in the field of neurobiology. Many of the neuronal substrates responsible for the effects that anesthetics are meant to subdue have been worked out in great detail. It has become far more apparent that the mechanisms involved in the normal transduction of human neural activity lie in interneuronal synaptic communications. Furthermore, synaptic transmission has been painstakingly characterized as the summation of electrical activity from a wide array of membrane spanning ion channel proteins. However, to date there has been no direct proof that one can alter a lipid bilayer (such as by dissolving an anesthetic in it) in such a way as to have subtle but direct effects on membrane spanning proteins as to grossly alter ion channel conductance, neuronal transmission and the subsequent behavior of a whole organism.
Direct Interactions of Anesthetics with Proteins
While hypotheses involving the direct effects of anesthetics on lipid bilayers indirectly affecting synaptic transmission have not been fully satisfactory, other investigations into the direct effect of anesthetics on proteins have proven more fruitful. Among the first to demonstrate the interaction of an anesthetic with a functional protein was the work of Franks and Leib.(9),(10) They demonstrated the direct enzymatic suppression of firefly luciferase, the enzyme responsible for the light of the firefly, upon the formation of the anesthetic-protein complex. While luciferase may not have anything to do with the anesthetic effect, this was the first work showing the alteration in protein function as a result of its direct interaction with an anesthetic.
Altered Anesthetic Modulation of Ion Channel Electrophysiology Via a Direct Effect
Many others have now gone on to show the electrophysiologic consequences of direct anesthetic binding to a variety of membrane-bound proteins actually associated with neuronal conductance. These include anesthetic effects on both voltage-gated (Na, K, Ca) and ligand-gated ion channels (glycine, GABA, AChR, serotonin, AMPA, kainate), as well as a variety of G-protein coupled receptors. In particular, the ligand-gated ion channels have been singled out because of their implication in the mediation of anesthetic-like states from other compounds (i.e., the benzodiazepine and barbiturate effects on GABAR), as well as several key studies demonstrating how they are fundamentally affected by the presence of anesthetics. Mihic et al.(11) first demonstrated the role of specific amino acids necessary for the anesthetic effect on the human glyRa1 in an oocyte preparation. They showed that mutation of serine 267 to a variety of other amino acids had significant bearing on the way anesthetics enhanced glycine-mediated chloride ion conductance.
The question that originally arose from this work was whether the effect of their mutations involved the actual anesthetic binding site or merely a related site that produced enough allosteric change to alter the actual anesthetic binding site. This issue was taken up by Mascia and colleagues(12) using a series of anesthetics derivatized with a methanethiosulfonate moiety. The same glyRa1 could be mutated from serine to cysteine at the same 267 position. This was shown not to significantly alter ion channel function. Then the mutated glyRa1 was exposed to the anesthetic propanethiol, which clearly produced an anesthetic effect on ion conductance that was reversible upon washout.
Next the mutated glyRa1 was exposed to propanethiol in the presence of reducing agents that would catalyze the formation of a covalent disulfide bond between the cysteine at position 267 and the propanethiol. This produced an anesthetic effect upon the ion channel that was of the same quality as that produced by the previous application of propanethiol alone, but it was irreversible even when all remaining reagent was removed from the system including the lipid bilayer. Both the same quality and magnitude of anesthetic effect was produced when the mutated system was exposed to propyl-MTS, the MTS derivative of the anesthetic propanethiol, forming the same disulfide bond more efficiently and without additional reducing agents. This demonstrated that, at least for the anesthetic propanethiol, it was both necessary and sufficient for the anesthetic to interact with the protein alone to produce its quantifiable effect. The mutated site was an actual anesthetic binding site and it was not necessary to implicate the lipid in the mechanism of action.
Finally Jurd and colleagues(13) have extended this result to demonstrate an effect on an entire organism. At the same homologous point in the mouse GABA beta 3 receptor, they produced a single point mutation that made mice completely insensitive to both propofol and etomidate, and partially insensitive to volatile anesthetics. While the result for the former two agents was quite dramatic, the effect of such a mutation on the modulatory capabilities of the volatile anesthetics was less well pronounced. In fact, the effect was only partial in nature at best, suggesting that the volatile anesthetics may have mechanisms that extend beyond a single ligand-gated ion channel effect, as noted above.
Studies of the Physicochemical Characteristics of an Anesthetic Binding Site
In order to better understand the possible physicochemical nature of the anesthetic binding site, many studies have been performed over the years that infer the possible characteristics of just such a site. Clearly the Meyer-Overton correlation still holds true for a great many anesthetic compounds, so the anesthetic binding site must have some hydrophobic character. The exceptions to the Meyer-Overton rule do reveal, however, a dependence on hydrophilic characteristics within the series of transitional compounds;(7) therefore, there must be a way to introduce hydrophilicity into the anesthetic binding site. Johansson and colleagues(3) advanced solubility studies by demonstrating anesthetic potency correlations within a series of solvents that simulated various amino acid side chain environments. Their work pointed to a methionine-like solvent as one that best mimics an anesthetic binding site. It should be pointed out that methionine is intimately involved at the GABA beta 3 site mutated by Jurd et al.(13) Several others have implicated weak hydrogen bond types of interactions within any anesthetic binding site.(14),(15) Trudell has clearly demonstrated the correlation of anesthetic potency with the polarizability of the noble gases in a polar environment.(16)
An Actual Anesthetic Site of Action: From Protein Schematic to Full- scale Atomic Model
Reasons to Study Molecular Mechanisms of Anesthetic Action
Consequently, our group has sought to better elucidate a more exact mechanism of anesthetic action on the molecular and atomic scale. Such a detailed mechanistic understanding should have multiple benefits. First, paramount to any future anesthetic drug design will be the understanding of ligand-target site interactions at an atomic level. Second, a more thorough knowledge of the consciousness-altering effects of anesthetics may further the cause of a greater insight into the actual neural, if not physicochemical, nature of consciousness itself. It is in this light that we have embarked on a two-pronged approach to studying the molecular mechanisms of anesthetic action. Our initial focus has been on the very detailed quantum mechanics analysis of anesthetic-protein interactions within several well characterized crystal structures of known atomic coordinates.(17) Because the proteins present in such currently available coordinate databases are simple globular structures not known to be involved in neural transmission, we have simultaneously used molecular graphics and modeling techniques to pursue our concurrent line of work: to build the first models of a putative anesthetic binding site within ligand-gated ion channels that are actually relevant to neuronal function.(18)
Physicochemical Basis of Anesthetic-protein Interactions
Most anesthetics are of a variety of shapes and sizes but are not very large molecules, so their overall binding motifs to proteins, in general, should be somewhat limited. In our initial studies, we set out to describe the general binding motifs of anesthetics to proteins in several well described anesthetic-protein complexes.(17) There have been several detailed studies of anesthetic-protein complexes that demonstrated direct binding of an anesthetic to a protein. They include bromoform-luciferase, halothane-albumin, halothane-cholesterol oxidase and dichloroethane-dehalogenase. Other less well defined crystallographic studies have included complexes of dichloroethane-insulin,(19) halothane-adeylate kinase,(20) halothane complexed with rhodopsin(21) and with various four helix bundles(22) that have been studied by spectroscopic means. Additionally, various azialcohol derivatives have been used to characterize anesthetic binding sites within both protein-kinase C(23) and nAChR.(24)
Based on our analysis of the aforementioned four high resolution crystal structures,(17) we have shown that the interaction of anesthetics with proteins involves a binding site characterized by several common hydrophobic and hydrophilic types of interactions. In fact, the binding pocket is amphipathic in nature, incorporating several anesthetic interactions with both a variety of amino acid types as well as water. Such interactions involve weak hydrogen bonds, van der Waals interactions and other weak polar interactions that many previous studies have implicated as necessary for reversible anesthetic binding. In particular, such an amphiphilic binding site actually induces significant polarization of the anesthetic itself, thereby contributing what may be a significant part of the anesthetic binding energy. These general binding motifs, when applied to ion channel proteins that may be responsible for anesthetic effects, may help to explain the relative potencies of the transitional compounds, as well as those that obey the Meyer-Overton correlation.
Such an amphiphilic binding site is also consistent with the work of several other groups.(25) Recent analyses of several halogenated volatile anesthetics have described the necessary combination of both "bulk" properties and electrostatic interactions to adequately account for anesthetic binding and potency. In particular, Sewell and Sear(26),(27) have used comparative molecular field analysis (CoMFA) to study the chemical characteristics of a large series of halogenated volatile anesthetics and have constructed an anesthetic pharmacophore that has both polar and nonpolar characteristics. This method is based solely on the analysis of ligands without reference to any type of specific protein binding site, yet reveals similar physicochemical characteristics for anesthetic binding.
Also, Abraham et al.(15) have suggested that "general anesthetic target sites in animals must have, in addition to their overall hydrophobicity, a polar component which is a relatively poor hydrogen bond donor, but which can accept a hydrogen bond about as well as water." Additionally, Sandorfy(14) has concluded that weak H-bonds and van der Waals contacts are the prime mediators of reversible anesthetic-protein interactions. As can be seen in the next section, our most recent work on constructing molecular models of anesthetic binding sites within the transmembrane four helix bundles of ligand-gated ion channels is highly consistent with this amphipathic notion.
Building Molecular Models of the Ligand-gated Ion Channels (LGICs) as Anesthetic Targets
Defining the Structural Problem
The initial schematic structure of the family of ligand-gated ion channels, as revealed by low resolution cryoelectron micrographs, was that of a three-component protein (Figure 1).(28)

Figure 1. Initial Schematic of Ligand-gated Ion Channel Structure.
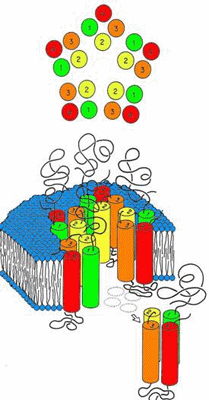
Modified from Olsen and Tobin, FASEBJ, 1990.
It was thought to be composed of a polar extracellular domain involved in native ligand binding, a hydrophobic transmembrane domain where mutations alter anesthetic modulation, and a somewhat mysterious intracellular domain involved in various cytoplasmic interactions. An ion channel pore traverses this entire protein for the passage of ions specific to the channel type. To form these regions, the overall schematic called for the axially symmetric placement of five protein subunits into a pentameric arrangement around the central ion pore (Figure 1). Within each subunit, the transmembrane domains seemed to be composed of four individual "rod-like" regions. The questions at this point were many. What is the secondary structure of the transmembrane "rods"? How are these transmembrane domains within the individual subunits arranged? How are the individual subunits themselves arranged to form a transmembrane ion channel? Where do the anesthetics have their effect?
For most proteins of interest, the typical sequence of study involves the purification of the protein material for crystallization and analysis by means of x-ray crystallography. This is especially important when one desires exact atomic coordinates for purposes of studying protein structure activity relationships and possible receptor-based drug design. However, the difficulty with the vast majority of membrane spanning proteins, especially the LGICs, lies in their denaturation or destruction of relevant protein structure when removed from their native transmembrane environment.
This has prompted our use of computer-based theoretical techniques with the incorporation of a variety of experimental restraints, so as to build realistic, physically relevant models of the ion channel proteins within the family of LGICs. Specifically, with modern techniques of bioinformatics, computational chemistry, and molecular modeling, we have built models of the transmembrane subunit region within human glycine alpha one receptors (GlyRa1) and gamma amino butyric acid receptors (GABAR) that account for a large body of the physicochemical and experimental data that characterizes these proteins. (18),(29),(30),(31),(32),(33),(34),
(35),(36) Most notably, these models contain cavities within the transmembrane region which demonstrate the proximity of amino acid residues known to be involved in the effects of anesthetics on these ion channels. This cavity forms a plausible anesthetic binding site that is amphipathic in physicochemical character and is large enough to contain an anesthetic.(17),(36)
Predicting Transmembrane Secondary Structure
Our initial work concerned the nature of the "rod-like" structures that composed the transmembrane tetrameric portion of the individual protein subunits.(34) To study this, the sequences of several human LGICs were obtained from the National Center for Biotechnology and Information. We utilized 10 state-of-the-art bioinformatics techniques to predict the transmembrane topology of this tetrameric region. The resulting sequences were aligned using the multiple sequence alignment method known as ClustalW, and the secondary structure predictions from each of the 10 topology prediction algorithms were superimposed onto the aligned amino acid sequences to analyze for consistency of prediction. Together, the consensus of these techniques predicted that the transmembrane subunits of the pentameric LGIC family were tetrameric bundles of alpha helices.
Building the Transmembrane Four-Helix Bundle and Discovery of an Anesthetic Binding Site
The next step was to use the principles of homology modeling in order to find a protein with a known three-dimensional structure and that looked in some way similar (either by sequence, protein fold or both) to the sequence and predicted fold of the LGICs.(18),(31),(33) Once such a protein of known structure was found, it could function as a template over which the sequence of the LGICs with unknown structure could be draped, with the subsequent transfer of three-dimensional coordinates to the new amino acid sequence. Therefore, the information regarding the alpha helical nature of the transmembrane rods was combined with the amino acid sequence information of various LGICs using the SeqFold search algorithm to search for modeling templates within a database of proteins with known three dimensional coordinates derived from the national Research Collaboratory for Structural Biology (RCSB, www.rcsb.org).
The SeqFold algorithm identified a relatively high-scoring modeling template in the coordinates of chain C of a bovine cytochrome oxidase (PDB ID 2OCC). This is a four-helix bundle of the up-down topology. The sequence of our LGIC was then aligned with this template using multiple scoring criteria. Refinement of the alignment closed gaps to produce agreement with experimental labeling studies that had been carried out by many groups on the homologous receptors of the superfamily. Since the large intracellular cytoplasmic loop between TMD 3 and 4 had little resemblance to any template, the model was then edited so as to contain an abbreviated version of the loop composed of only 9 native residues from the loop region itself along with a chain of 5 glycine residues substituted for the central remaining bulk of the loop.
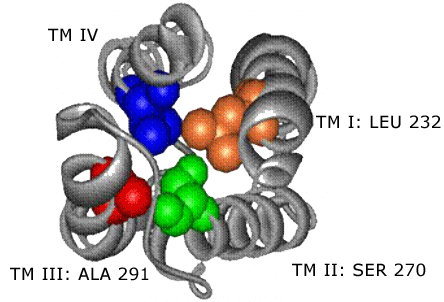
Structural assignment and refinement was achieved using the Modeler program. The final structure demonstrated a cavity within the core of a four-helix bundle. Residues known to be involved in modulating anesthetic potency converge on and line this cavity. This suggests that the binding sites for volatile anesthetics in the LGIC's are the cavities formed within the core of transmembrane four-helix bundles (Figure 2).
Figure 2. The Structure of the Four-helix Bundle Transmembrane Domain.

Four alpha hlices
Building the Entire Transmembrane Domain
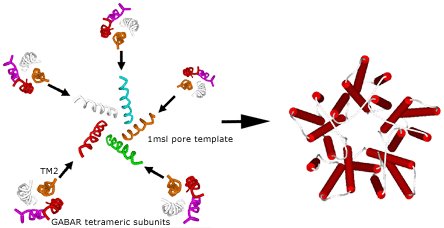
Once the four-helix bundle nature of the individual transmembrane domains had been identified, the subunit models had to be combined into their pentameric arrangement, so as to produce the entire transmembrane domain of the ion channel. For this purpose a new template, for a different form of alignment involving only the pore lining helices, was found in the analogous bacterial mechanosensitive channel (PDB ID 1MSL). Only the channel-lining helices were used as the pentameric template around which to organize the channel-lining helices of our LGIC tetrameric subunit, so as to build a pentamer of tetramers for the entire TMD construct (Figure 3).
Figure 3. Merging the Subunits to Form the Entire Transmembrane Domain.
Building the Ligand-binding Domain (LBD)
For the extracellular LBD, a homology model was built based upon the acetylcholine binding protein (AChBP, PDB ID 1I9B) of Sixma and coworkers(37). Here, the sequence of a given LGIC was draped over the AChBP template with known 3D coordinates and energy optimized. In particular, the heteromeric GABAR is composed of 2 alpha subunits, 2 beta subunits and 1 gamma subunit. The determination of the unique clockwise orientation of these subunits around the central ion pore was made possible for the first time by the alignment of specific residues known to bind various ligands within the extracellular domain, most notably those residues involved in benzodiazepine binding. This clockwise orientation arranged the subunits in the order of gamma, alpha, beta, alpha, beta around the ion channel pore.
Aligning and Merging the TMD to the LBD
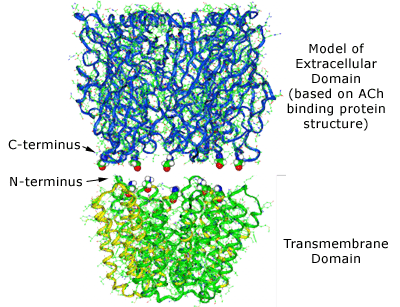
The LBD and TMD were then mated (Figure 4), according to distance restraints imposed by mutational data, with a surprisingly good fit and thereby producing the entire LGIC complex minus its intracellular cytoplasmic domain.(35),(36)
Figure 4. The Combination of the Extracellular Ligand-binding Domain with the Transmembrane Domain.
↓

Note the excellent tapered fit based on both outside protein perimeters and pore lining walls. Note also the contiguous ion channel pore traversing the entire protein complex.
Using the various force fields of molecular mechanics, hydrogens were then added to the homology model structure and an automated conformation check performed to obtain a low energy state of the amino acid side-chains. After initially tethering the protein backbone, the entire complex underwent a series of restrained optimizations to eliminate any additional high-energy conformations. In the end, final energy optimization was obtained without restraints and without gross distortion of tertiary structure.
Results and Analysis of the LGIC Model
Our current models of human glyRa1 and GABARa1 homomers have demonstrated several key findings. The extracellular LBD has a large component of beta sheet structure between the subunits of which native ligand binding may occur. This LBD mates extremely well with a model of the TMD built by completely independent means. The transmembrane domain is composed of a pentamer of tetrameric alpha helical bundles, within which lies a putative anesthetic binding site. This site illustrates the convergence of residues known to be involved in anesthetic effects on these ion channels.(11),(32) The size of the pore channel is in a range large enough to accommodate ion passage. The residues involved in the ion selectivity filter are located around the distal end of the TMD pore.(38) While our model was developed from completely theoretical means, the overall protein construct can be seen as extremely similar to the most recent model of the nicotinic acetycholine receptor (2BG9.pdb) within torpedo californicus derived from the newly revised cryoelectron microscopy of Nigel Unwin (Figure 5).(39)
Figure 5. The Top View (A) and Side View (B) of Our Current Model of a Ligand-gated Ion Channel.
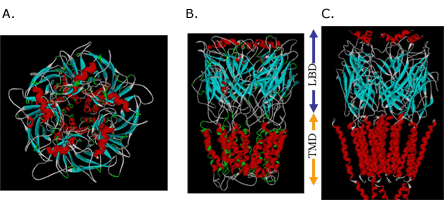
Note the similarity to the intermediate resolution cryoelectron micrograph of Unwin (C).
How the LGIC Moves: The Results of Normal Mode Analyses and Its Implications
With our receptor construct now relatively whole and a putative anesthetic binding site identified, it has been our eventual goal to seek how the binding of anesthetics and other ligands may alter overall ion channel function. Current computational capabilities could allow us to perform formal molecular dynamics simulations on a relatively small protein for the further examination of larger scale motions in exacting detail. However, to perform large scale molecular dynamics calculations for the millisecond timescale that is required for actual ion channel opening and closing on our LGIC models that contain approximately 26,000 atoms each, is intractable even with the most modern of computational hardware.
We have therefore resorted to the more approximate techniques of normal mode analysis to study large scale protein motion.(40),(41) Normal mode analysis is a computational means of decomposing the whole motion of a protein into individual orthogonal vibrations. We had hypothesized that the highest amplitude, lowest frequency normal mode of the protein should characterize the natural resonance of channel gating within a LGIC. That is, the natural opening and closing motion involved with ion passage should be represented in the highest amplitude harmonic vibration of the protein. We performed these calculations using three different methods of normal mode analysis and our hypothesis proved to be correct.These calculations demonstrate a great many characteristics of LGIC motion and have many implications for future study. It can be shown that one of the most fundamental motions of our LGIC models is characterized by an "iris-like" wringing motion, as one might wring water from a washcloth in opposite directions when gripping from opposite ends (Figure 6). This motion produces an alteration in pore diameter in the general direction of ion channel opening. The frequency of such motion appears to occur on the subnanosecond time scale. While this seems a bit faster than the experimentally derived time constant for ion channel gating, this time scale implies that the ion channel is vibrating in the general direction of channel gating on a virtually continuous basis. However, it is probably not open long enough, on the average, for actual ion transduction. It would not be until a random thermal event occurred that was high enough in energy to cause a large enough change in amplitude or time spent in the open state that spontaneous channel opening would occur.
A plot of the backbone deviations during motion of the residues known to modulate anesthetic effect when mutated demonstrates that the putative anesthetic binding pocket seems to be consistently located in a region of intermediate motion. This may prove to be some critically important "hinge point" within the complex. This cavity may also have a transmembrane/boundary mode of access. Anesthetic binding may actually alter the overall motion of a ligand-gated ion channel by a "foot-in-door" motif, resulting in the higher likelihood of and greater time spent in the open channel state. The transduction of native ligand binding from the extracellular LBD to the TMD occurs via two methods of coupling: The first method is by way of the protein backbone, as the amino acid sequence transitions from the LBD to the TMD. The second is through the intercalation of the TMD2-3 loop with loops 2 and 7 from the LBD.
Conclusion and Future Directions
For the last one and a half decades, anesthetics have been given both safely and effectively for the relief of patient suffering. While a great many theories have been put forth, it has become increasingly apparent that a significant focus of anesthetic action lies within the family of ligand-gated ion channels (LGICs).
These channels have a transmembrane region that is composed of a pentamer of four helix bundles within which lies a putative amphipathic anesthetic binding site. This amphiphilic nature may help to explain the need for hydrophobicity expressed in the Meyer-Overton correlation, as well as the need for hydrophilicity required by the exceptions to the Meyer-Overton rule.
The overall motion of these channels is consistent with the natural harmonic vibrations demonstrated by normal mode analyses. While still the goal of future pursuits, anesthetic binding may alter the overall motion of a ligand-gated ion channel by a "foot-in-door" motif, resulting in the altered likelihood of ion channel conductance in the presence of native ligand. Despite the great distance from the native ligand binding site to the gating elements of the pore, such analysis also demonstrates a ready means of information transduction over a large span. In the future, such calculations may allow for the analysis of subtle effects from mutations and small ligand binding. In addition, such analyses may also allow the use of protein dynamics, elucidated via normal mode calculations, as additional endpoints for future drug design.
Figure 6a. Glyra - Side View.
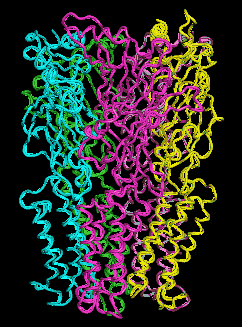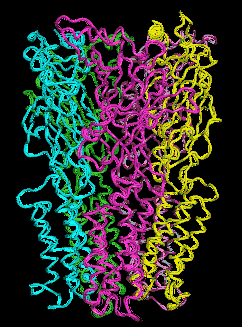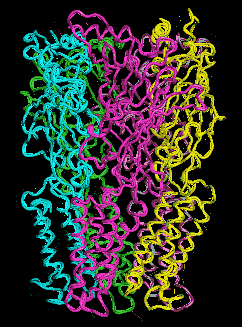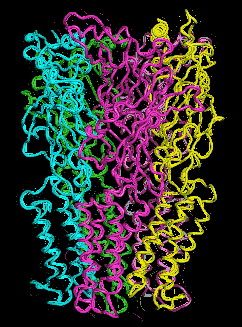
Figure 6b. Glyra - Top View.