Course Authors
Stephen R. Thom, M.D., Ph.D.
Release Date: 07/31/2007
Upon completion of this Cyberounds®, you should be able to:
Discuss the overlapping elements associated with CO pathology
Discuss the latest information on clinical evaluation and health risks for CO poisoning
List the CO poisoning treatment options, including evidence related to hyperbaric oxygen therapy (HBOT).
Though emergency physicians are aware of the general risks posed by carbon monoxide (CO), new research findings are beginning to influence clinical management. With this new information about the cardiac and nervous system risks from CO and the underlying mechanisms for injury, it is hoped that the high incidence of morbidity from CO can one day be reduced and that the clinical management of CO poisoning will become less challenging.
Environmental CO contamination from incomplete combustion of carbon-containing substances is a major public health threat. Many poisonings could be avoided simply by improved communication about its dangers. This is an international problem and CO may be responsible for over half of all fatal poisonings.(1),(2),(3) It is estimated that CO poisoning is the third leading cause of unintentional deaths in the United States and that the incidence of nonfatal poisonings varies from 15,000 to 40,000 cases per year.(1),(2),(3),(4),(5),(6),(7) Because misdiagnosis of CO poisoning is common, however, the true incidence is likely much higher.(8),(9)
Pathophysiology
The affinity of CO for heme-containing proteins is well-established -- five processes occur simultaneously and contribute to CO poisoning (Table 1). Below is a brief discussion of each process.

Table 1. Mechanisms of CO Pathology.
|
Impaired O2 Delivery and Utilization
CO has a high affinity for binding to hemoproteins and deleterious effects can occur as the result of impaired O2 delivery. The affinity of CO for hemoglobin is more than 200-fold greater than that of O2 and formation of carboxyhemoglobin (COHb) is well-recognized as an effect of CO exposure.(10) When CO occupies hemoglobin binding sites for O2, the arterial O2 content is reduced. CO binding also influences the sigmoidal shape of the oxyhemoglobin dissociation curve, interfering with the release of O2 to the tissues. The 'left' shift of the oxyhemoglobin dissociation curve results in an exaggerated lowering of venous O2 partial pressure (Figure 1).(11)
Figure 1. Oxygen-hemoglobin Curve.
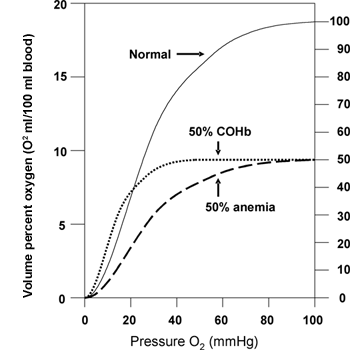
Three curves are shown: One (solid line) depicts the normal dissociation curve, the dashed line shows the impact of reducing the hemoglobin content by half (anemia) and one (small dotted line) the impact of 50% COHb. The left shift associated with COHb demonstrates the extra adverse effect of CO versus merely loss of hemoglobin O2 carrying capacity. The partial pressure of O2 when hemoglobin gives up 50% of its available O2 is at ~16 mm Hg when COHb is 50%, versus ~26 mm Hg when there is a 50% anemia.
CO perturbs cellular bioenergetics because it inhibits mitochondrial respiration by binding to cytochrome oxidase.(12),(13) CO only binds to the reduced form of the enzyme, however, and experimental observations with respiring tissues have shown that the CO concentration must be 12- to 20-fold higher than that of O2 to reduce mitochondrial O2 uptake by 50%.(14),(15) In the brain, in addition to impeding ATP synthesis, CO binding to mitochondrial hemoproteins leads to the production of reactive O2 species.(16),(19),(20),(21)
O2-CO-nitric oxide (·NO) Competition
Intracellular hemoprotein functions are influenced by the partial pressures of the various protein ligands: O2, CO and ·NO. These gases bind competitively to hemoproteins so their effects depend on relative concentrations. This is clinically important because exogenous CO will perturb the equilibrium concentration among the ligands. CO disturbs the association between ·NO and hemoproteins so that the steady state concentration of ·NO is elevated in, and around, both platelets and endothelial cells.(22),(23),(24) Toxic effects occur because the liberated ·NO is available to undergo alternative reactions.
Perivascular Oxidative Changes
Platelet-neutrophil aggregates and intravascular neutrophil activation occur in association with CO poisoning.(25) The ·NO liberated from CO-exposed platelets reacts with superoxide anions (O2·) generated by nearby neutrophils in the blood to produce reactive species that trigger aggregation. Once a physical linkage between platelets and neutrophils is established, neutrophils exhibit a marked increase in oxidative burst, synthesis of additional reactive ·NO-derived species, and the neutrophils undergo degranulation.(26) Platelet-neutrophil aggregates are found in CO-poisoned animals and in patients.(25) Myeloperoxidase (MPO) liberated from the neutrophil primary granules is also deposited along vascular walls, causing endothelial cell oxidative stress.(25),(27),(28),(29),(30),(31),(32)
Excitotoxicity
Elevations in excitatory neurotransmitters occur in the brain during CO poisoning.(21),(33),(34),(35) This could be caused by any (or all) of the three components of CO pathology already listed. Toxic products, especially nitrite, and also alterations of intracellular calcium ions exacerbate neuronal stress/injury.(36)
Immunological Responses
Perivascular oxidative stress occurs as a consequence of the four components discussed above. Reactive chemical species are generated by these events and by perivascular enzymes that become activated. These species react with and modify the three-dimensional structure of brain myelin basic protein (MBP). The immune system responds to the altered MBP and subsequent brain inflammation precipitates neurological damage.(32)
Clinical Presentation and Evaluation
Signs and Symptoms
Initial symptoms arising from CO exposure are headache, nausea and dizziness. This presentation mimics many common conditions, which can delay diagnosis and identification of CO environmental contamination.(9),(37),(38),(39),(40),(41),(42),(43),(44),(45) Common physical findings include tachycardia and tachypnea; blood pressure changes are variable.(46) Dysrhythmias, myocardial infarctions and sudden cardiac arrest are reported in response to CO poisoning.(47),(48),(49),(50)
Clinical observations and animal trials indicate that acute mortality from CO poisoning is probably related to cardiac injuries. Some of the cardiac damage arises from hypoxic/ischemic stress, particularly in patients with underlying coronary artery disease.(47),(48),(51),(52) Recent studies have identified a high incidence of more global cardiac insults. Animals exposed to modest CO levels, where the COHb is on the order of 11%, exhibit an increase in coronary perfusion pressure and impaired contractility that lasts for 48 hours.(53) Recent clinical reports have described a high incidence of cardiac injuries in moderate to severely CO poisoned patients with normal coronary arteries.(54),(55)
Impaired mitochondrial respiration is a possible mechanism for the cardiac insult from CO, although this should not persist after removal from the CO environment. Insults mediated by platelet-neutrophil interactions offer an alternative mechanism, though not yet proven. Platelet-neutrophil interactions, reduced neutrophil MPO index (MPO/cell) and elevated intravascular MPO are linked to a heightened risk for acute coronary syndromes.(56),(57),(58) A rare physical finding often quoted in the older literature is cherry red coloration of the skin. Superficial blisters in dependent areas can be found when patients have been laying comatose for a period of time but these are probably a non-specific finding.(59),(60)
Laboratory Testing
Measurement of COHb by spectrophotometry is the standard method for confirming the diagnosis of CO exposure. Normal levels in non-smokers are between 0.2 and 0.85%. Smokers often have COHb levels of around 4% and with heavy smoking, 10% COHb. In the evaluation of neonates, it is important to note that absorption characteristics of fetal hemoglobin are close to those for COHb and this may cause confusion.(61) Pulse oximetry is an unreliable method for estimating CO exposure because most instruments cannot discern the difference in spectral characteristics between oxyhemoglobin and COHb.(61)
Because of the risk of cardiac injuries from CO, obtaining an EKG and screening plasma biomarkers indicative of injury are prudent. This matter has been under-appreciated, as those who suffer an acute cardiac injury have an increased risk for cardiovascular-related death in the following 10 years.(55) A chest X-ray also should be part of the emergency evaluation. This is an obvious point in patients with concurrent smoke inhalation; however, pulmonary vascular congestion and alveolar infiltrates can occur with isolated exposures to CO secondary to compromised myocardial function.(62),(63)
A correlation has been reported between COHb level and symptoms of headache and dizziness among patients with relatively low COHb levels on the order of 2 to 10%.(41),(42) Unfortunately, no reliable correlation exists between COHb level and more severe signs and symptoms. Absence of objective measures for establishing the severity of CO poisoning remains among the most troublesome aspects of clinical evaluations. Newer tests such as measurements of plasma myeloperoxidase (MPO) and platelet-neutrophil aggregates have not yet been correlated with poisoning severity or prognosis in prospective trials.(25)
Neuroimaging
Many clinical reports have documented brain computed tomography (CT) and magnetic resonance (MR) image abnormalities following CO poisoning.(64),(65),(66),(67) Neuronal insults mediated by CO are not anatomically discrete. Imaging studies suggest that CO neurotoxicity can, consistent with animal studies, involve a vascular injury. White matter changes may be found in approximately one-third of patients with severe poisoning. Lesions in the centrum semiovale are related to worse cognitive outcomes.(71) Unless there is concern for a concurrent intracranial bleed, however, neuroimaging in the ED is not likely to assist with acute evaluation of the CO poisoned patient.
Treatment
Emergency Stabilization
Initial intervention should follow standard management practices with the provision of a patent airway and support of circulation. Administration of supplemental O2 is the cornerstone of treatment for CO poisoning. Oxygen inhalation will hasten dissociation of CO from hemoglobin, as well as provide enhanced tissue oxygenation. There is reasonable agreement among studies in human beings that the mean COHb half-life while breathing air is near 320 minutes but measured values in individual studies vary from 128 to 409 minutes.(37),(72),(73),(74),(75),(76),(77) In patients breathing 100% high flow O2 by mask, the average half-life is approximately 113 minutes but measured values for individual patients span from 27 to 464 minutes.(37),(75),(77) Because of such broad individual variations, the attempt to extrapolate and estimate a patient's COHb at the time of ED evaluation is a futile exercise.
The reduction of COHb half-life by O2 has historical importance to the field of hyperbaric oxygen therapy (HBOT), as the notion of hastening COHb removal was the initial impetus for the consideration of this treatment. The mean COHb half-life decreases to approximately 23 minutes when breathing O2 at 3 atmospheres absolute (ATA).(72) In an ironic twist, however, animal studies indicate that the efficacy of HBOT is probably not based on this action. In fact, an animal trial suggests that HBOT does not reduce neurological injury mediated by CO-induced hypoxia.(78)
Actions of HBOT, but not ambient pressure O2 treatment, that have been demonstrated in animal models to ameliorate pathological events include an improvement in mitochondrial oxidative processes,(79) inhibition of lipid peroxidation(80) and impairment of leukocyte adhesion to injured microvasculature.(81) Animals poisoned with CO and treated with HBOT have been found to have more rapid improvement in cardiovascular status,(82) lower mortality(83) and lower incidence of immune-mediated neurological sequelae.(84)
Clinical Trials of HBOT
Published clinical trials span 18 years and a broad range in quality. Efficacy of HBOT for acute CO poisoning is well supported in animal trials and recent studies provide a mechanistic basis for treatment.(84) In this era of evidence-based medicine, a great deal of emphasis has been placed on systematic reviews. The fidelity of reviewers' analyses has a profound impact on these assessments, as was reflected in online communications about a recent Cochrane Review.(85)
The clinical efficacy of HBOT for acute CO poisoning has been assessed in five prospective, randomized trials published in peer-reviewed journals.(86),(87),(88),(89),(90) There is only one clinical trial by Weaver et al. that satisfies all items deemed to be necessary for the highest quality trials.(91) This study reported a randomized, double-blinded, placebo-controlled human clinical trial involving 152 patients.(90) All enrolled patients received treatment with either three sessions of HBOT therapy or normobaric O2 with sham pressurization to maintain blinding. Critically ill patients were included, with half of enrolled patients having lost consciousness and 8% requiring intubation. The follow-up rate was 95% with assessments performed by trained examiners and compared with age, sex and education-controlled norms.
The definition of neurological sequelae, defined a priori, was fulfilled in symptomatic patients by an aggregate performance on six neuropsychological tests that was at least one standard deviation below predicted, or by an aggregate score of two or more standard deviations below expected in asymptomatic individuals.
At 6-weeks post-poisoning, the cognitive sequelae rate was 25% in subjects treated with HBOT compared with 46% of patients treated with normobaric oxygen (P=0.007). When adjusted for cerebellar dysfunction and stratification, the odds ratio was 0.45 (p=0.03; 95% CI=0.129-0.919). At one year, 33% of patients not treated with HBOT had cognitive sequelae versus just 18% if treated with HBOT.
Chronologically, the next most recent trial by Scheinkestel et al.(87) reported on 191 patients treated with continuous O2 by face mask for three days after CO poisoning with daily trips to the HBOT chamber. Patients with severe poisoning were included and more than half were comatose. In order to maintain blinding, patients randomized to the non-HBOT group received "sham" pressure treatments. Additional treatments (up to six daily sessions) were performed in patients without neurological recovery. The primary outcome measure for this trial was testing performed at completion of treatment (3-6 days) and not from long-term follow-up.
The Scheinkestel study had a high rate of adverse neurological outcomes in all patients, regardless of treatment assignment. Neurological sequelae were reported in 74% in HBOT-treated patients and 68% in controls. No other clinical trial has approached this degree of neurological dysfunction. The high incidence is likely related to the assessment tool which could not discern true neurological impairments from poor test-taking secondary to depression.(92) Suicide attempts with CO represented 69% of cases in this trial. Outcomes at one-month were not reported but remarked to show no difference. Unfortunately, there were 54% of subjects lost to follow-up. Multiple statistical comparisons were reported without apparent planning or statistical correction. Both treatment arms received continuous supplemental mask O2 for three days between their hyperbaric treatments (both true HBOT and 'sham'), which resulted in greater overall O2 doses than conventional therapy. Flaws in the design and execution of this study make it impossible to draw meaningful conclusions from the data.
Thom et al.(89) reported a benefit to HBOT in a study of 65 CO-poisoned patients randomized to a single HBO2 treatment session or mask O2. Patients in this trial suffered from mild to moderate poisoning, as those with loss of consciousness were not included, and blinding was not employed. The primary outcome measure (self-reported symptoms of neurological sequelae combined with deterioration in at least one of six neuropsychological tests occurring at any time after treatment) was found in 0% (95% CI 0% to12%) of the HBOT-treated patients and 23% (95% CI 10% to 42%) of the patients treated with ambient pressure O2. All patients with reported neurological sequelae had resolution by 77 days. Limitations in this trial were lack of blinding and selection of a subgroup of patients likely to have suffered less severe poisoning.
A prospective trial by Ducasse et al. randomized 26 patients with acute CO poisoning to receive normobaric O2 (100% oxygen for 6 hours, followed by 50% oxygen for 6 hours) or HBOT (one 2-hour treatment, followed by 4 hours of 100% normobaric O2, followed by 6 hours of 50% normobaric O2).(86) Poisoning was accompanied by loss of consciousness in 65% of the patients. Outcome measures included symptoms, electroencephalogram and cerebral blood flow responses to acetazolamide administration. A significant benefit at three weeks was seen in the HBOT treatment group (p<0.02). Limitations of this trial included small size, an inadequate randomization procedure so that treatment allocation was not appropriately concealed and the use of surrogate outcome measures.
Raphael et al.(88) studied 343 CO poisoning patients without loss of consciousness who were randomized to one HBOT session or an equivalent duration of mask O2. This was an un-blinded trial and the primary outcome measure was a symptom questionnaire, supplemented by physical and neurological examination, in an unspecified number of patients. One month after treatment, 32.2% of patients who received HBOT and 33.8% of control patients reported neurological symptoms (P=0.75, NS, chi-squared), and 97% of patients in each group had resumed their previous occupation. Data from this study were republished with additional subgroup analysis showing no change in outcome.(93) This study has been criticized for using overly broad inclusion criteria, an inadequate regimen for HBOT, long treatment delays and weak outcome measures.(94),(95)
If treatment decisions are based on the highest quality trial by Weaver et al., their post hoc subgroup analysis offers guidelines for the use of HBOT therapy. HBOT reduced cognitive sequelae in patients with any of the following: loss of consciousness, COHb >25%, age >50 years or a base excess <-2 mEq/L. In patients with none of these criteria, HBO2 did not improve outcome. The preponderance of evidence indicates that HBOT treatment significantly reduces the incidence of neurological sequelae and, in retrospective comparisons, appears to also diminish acute mortality.(96) As yet, however, there has not been an assessment as to the length of delay from poisoning beyond which there is no chance for benefit from HBOT.
Maternal-fetal CO Poisoning
Maternal CO poisoning is a special situation that deserves additional commentary. Maternal symptoms at the time of exposure more closely predict the risk of associated fetal morbidity/mortality than COHb.(97) Severe CO poisoning is associated with a maternal mortality between 19% and 24% and a fetal mortality between 36% and 67%.(98) When mother and fetus survive, many fetuses subsequently developed somatic and neurologic sequelae, including limb malformations, microcephaly, hypotonia, areflexia, persistent seizures, mental and motor disabilities.(99),(100)
Hypoxic stress related to impaired O2 delivery is an obvious component of fetal distress. Normal fetal arterial pO2 is low, about 20 mm Hg versus 100 mm Hg for maternal arterial blood. Hence, the fetal O2 exchange typically occurs near the steep part of the oxyhemoglobin dissociation curve. A small drop in maternal pO2 can cause a precipitous drop in fetal pO2. This physiological stress occurs more quickly than that associated with CO binding to fetal proteins. Studies with sheep demonstrated that fetal COHb does not reach steady state until approximately 36 to 48 hours, whereas maternal COHb reaches steady state in 7 to 8 hours.(101)
The second insult related to fetal COHb is a disturbance in the O2-hemoglobin dissociation curve. Binding by CO causes a left shift of the curve, which increases the hypoxic stress to the fetus. Fetal COHb concentration rises more slowly than does maternal COHb but once steady state is reached the fetal level is higher. The increased level is related to the higher affinity fetal hemoglobin has for CO as compared with hemoglobin A.
The human fetal-maternal COHb concentration ratio is 1.1 to 1.0.(102) That is, at steady state the fetal COHb concentration will be 10 to 15% higher than maternal COHb. While the slow kinetics may be viewed as a protective factor for the fetus, the dynamics work in reverse for CO elimination. The half-life for fetal COHb is nearly twice that for maternal COHb.(101) Therefore, there is a physiological basis for treating CO poisoning in pregnant women with ambient pressure O2 for longer than the time it takes to register negligible maternal COHb. Whether complex intravascular processes such as CO-induced platelet-neutrophil aggregation occur in the fetal circulation are unknown.
Anecdotal clinical reports suggest that HBOT may improve fetal outcome.(98),(103),(104),(105),(106),(107),(108) The only experimental study that addresses the efficacy of HBOT for reducing fetal risk from acute CO poisoning showed a reduction in spontaneous abortion in pregnant rats.(109) There are no significant extra risks presented to the fetus or mother from HBOT if therapeutic protocols are followed.(110),(111) The current recommendations for use of HBOT in pregnant women are the same as those for any other patient.
Summary
There persists a common misconception that the risks posed by CO are all secondary to hypoxic stress from hemoglobin binding. This review has outlined additional threats. Indeed, these alternative mechanisms are likely more important for emergency physicians because morbidity from CO is much more frequent that mortality. CO threatens life because of cardiac decompensation. Neurological injuries arise from hypoxia and a complex interplay involving perivascular oxidative stress and excitotoxicity.
The 'auto'-immune events that evolve in the days/weeks that follow acute poisoning can be avoided by prompt intervention. There are several theoretical treatments that might benefit patients but the only treatment proven to be effective by basic science/animal studies and in randomized clinical trials is hyperbaric oxygen. Of late, it has become clear that one pays a political price for making this statement but the data are quite clear. It is also clear that the best intervention is education, as the only way to decrease the alarmingly high incidence of poisoning is by improving awareness of the danger. Efforts are underway to identify objective markers for patients who have suffered high-risk poisonings. In the meantime, the best management of patients in day-to-day emergency practice requires a high index of suspicion, obtaining a careful history and judicious use of laboratory tests including markers for cardiac injury, as well as COHb.