Course Authors
Rodney M. Camire, Ph.D.
Release Date: 04/16/2007
Upon completion of this Cyberounds®, you should be able to:
Discuss the role of factor VIII and factor IX in hemostasis
Describe the basic elements of the factor VIII and factor IX genes
Describe the underlying genetic causes of hemophilia
Describe currently available treatment regimens for hemophilia
Describe future therapeutic strategies to treat hemophilia A and B.
Normal hemostasis requires adequate levels and function of soluble blood coagulation proteins, platelets and vessel wall components. These constituents act in concert to initiate a focal and measured response to vascular damage which ultimately results in the formation of a blood clot. The major proteins involved in the formation of thrombin, the enzyme ultimately responsible for fibrin formation and platelet activation, are shown in Figure 1.

Figure 1. The Coagulation Response.
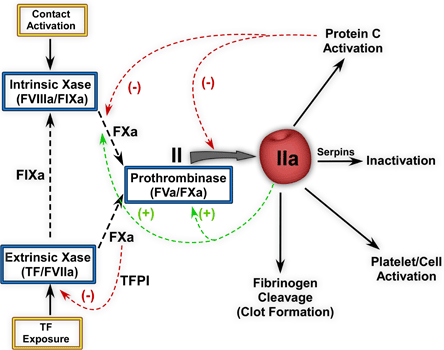
Tissue factor (TF) exposure leads to the formation of the extrinsic Xase complex and initiates the coagulation response. This enzyme complex activates FX to FXa and can also activate FIX to FIXa. Once extrinsic Xase is downregulated by TFPI, the intrinsic pathway sustains FXa generation and ultimately thrombin generation. Prothrombinase is responsible for the formation of thrombin (IIa). Red arrows indicate negative feedback reactions while the green arrows represent positive feedback reactions.(2)
Hereditary defects in any of these proteins may result in a bleeding diathesis. Classical hemophilia is a bleeding disorder resulting from a deficiency of factor VIII (hemophilia A) or factor IX (hemophilia B). These proteins are involved in the intrinsic pathway of coagulation. Hemophilia A and B are the most frequent bleeding disorders and, together with von Willebrand disease, account for ~95% of all inherited deficiencies of coagulation factors.
The factor VIII and IX genes are located on the X-chromosome; thus their deficiency is classified as an X-linked recessive bleeding disorder, which almost exclusively affects males. Deficiencies in other coagulation factors (i.e., factors X, VII, V, etc.) can also result in bleeding but are not typically classified as "hemophilia". Rather, they are classified as "rare bleeding disorders" and are inherited as autosomal recessive traits affecting ~1:1,000,000 individuals.(1)
The focus of this Cyberounds® review is to describe the molecular genetics of classical hemophilia (hemophilia A and B), briefly discuss current treatment options and also introduce to the reader new therapeutic strategies in development for treating these disorders. To assist the reader in finding information about the factor VIII and factor IX genes, Table 1 provides their gene symbols, identification numbers for various databases as well as other useful gene information.
Factor VIII Deficiency (Hemophilia A)
Factor VIII is a 330 kDa glycoprotein consisting of a variably sized heavy chain and a light chain which are held together by divalent metal ions (Figure 2). The heterogeneity of the heavy chain (90-200 kDa) is due to variable intracellular processing of the central B domain.
Figure 2. Schematic Representation of FVIII and FVIIIa.
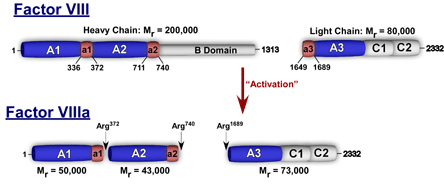
Boundaries of the acidic regions denoted by a1, a2, and a3 are indicated.
Factor VIII has multiple domains (A1-A2-B-A3-C1-C2) and shares homology with factor V. It is synthesized in the liver and circulates in plasma at a relatively low concentration of ~100-200 ng/mL (0.3-0.6 nM) with its carrier protein von Willebrand factor (vWF). In order for factor VIII to properly function in the intrinsic pathway, it must be proteolytically processed by thrombin or factor Xa. The resulting activated factor VIII (FVIIIa) is a heterotrimer and functions as a cofactor protein for factor IXa in the intrinsic Xase complex. This enzyme complex is the physiological activator of factor X.(3) The essential role of factor VIII in blood coagulation is evidenced by the severe bleeding diathesis associated with its deficiency (hemophilia A) which occurs with a frequency of ~1 in 5000 males.(4) A mouse model for hemophilia A has been established using gene knockout technology and is available for research use.(5)
The factor VIII gene was originally cloned in 1984 and is ~186 kb in length, has 26 exons and lies on the X chromosome (q28).(6),(7),(8) The gene encodes a mature mRNA of ~9 kb and the cDNA is 7056 nucleotides long.(9),(10)The first description of mutations in the factor VIII gene dates to 1985. In this paper by Gitschier et al., two nonsense mutations in the coding sequence and two partial deletions of the factor VIII gene were described.(11) The finding of four distinctly different mutations in the factor VIII gene of otherwise similar patients was the first hint that a large variety of mutations exist in the factor VIII gene in hemophilia patients. This prediction was borne out after PCR was introduced for large-scale point mutation detection.
A common rearrangement in the FVIII gene discovered in 1993 is an inversion that is highly prevalent among severe hemophilia A patients.(12),
The molecular pathology of the factor VIII gene is constantly being updated and reviewed in an electronic version of the hemophilia A database:
http://europium.csc.mrc.ac.uk/WebPages/Main/main.htm(16).
In 2003, there were more than 940 unique mutations in factor VIII of all types reported. More than 600 different point mutations are known for the factor VIII gene, and this number is increasing steadily as more patients are analyzed. The effect of these mutations on gene function is diverse. Severe dysfunction is produced by point mutations that introduce premature stop codons and by mutations that destroy splice junctions between introns and exons. On the other hand, these types of mutations also contain most, if not all, of the mild and moderately severe forms of hemophilia A. These are mostly missense mutations that predict replacement of the normal amino acid by another amino acid. These changes to FVIII can influence: synthesis, processing or secretion of FVIII; binding to vWF; activation by thrombin; stability; phospholipid binding; and binding to FIXa.(17)
Gene deletions of less than 100 bp up to several hundred kilobases (kb) have been found in many severely affected patients. In fact, deletions may be present in almost 5% of severe hemophilia A cases.(18) This estimate is somewhat biased in that gross gene arrangements are readily detected by a simple Southern blot, whereas point mutation detection requires elaborate screening of all the coding regions of the factor VIII gene.(19)
Insertions are relatively uncommon in the factor VIII gene.(16) As with deletions, it is important in principle to distinguish in-frame from frameshift insertions. It should be noted, however, that the insertions known to date have always been found in severe patients with undetectable levels of factor VIII activity. In the group of large insertions are the so-called LINE (long inserted element) retrotransposons.(20),(21) These DNA elements comprise approximately 5% of the human genome. Intact LINE elements encode a protein with reverse transcriptase-like activity and have the capacity to spread through the genome through an RNA intermediate. The LINE element first identified in hemophilia A patients was derived from an element on chromosome.(22) Subsequent to the original description in hemophilia A, insertion of LINE elements has been found to disrupt various other genes.
Factor IX Deficiency (Hemophilia B)
Factor IX is a vitamin K-dependent, serine protease zymogen that plays a critical role in the intrinsic pathway of blood coagulation. Factor IX is primarily synthesized in the liver and is comprised of 461 amino acids (pre-pro-factor IX; Figure 3).
Figure 3. Processing of Factor IX.
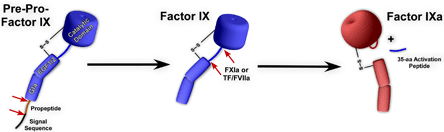
Factor IX is synthesized with a signal sequence and propeptide which are removed prior to secretion. Factor IX is a zymogen and has no enzymatic activity. Factor IXa is generated following proteolytic removal of a 35-amino acid activation peptide by factor XIa or tissue factor VIIa.
Following proteolytic processing and removal of the signal sequence and propeptide, it circulates in plasma as a single chain 415 amino acid protein (55 kDa) at a concentration of ~5 μg/mL (90 nM). Factor IX is activated to factor IXa by factor XIa or tissue factor/factor VIIa following cleavage at two sites (Figure 3).(22) Activated factor IX is the serine protease component of the intrinsic Xase enzymatic complex, which is also comprised of factor VIIIa, anionic membranes and calcium ions.
The factor IX gene was cloned between 1982 and 1984 and lies on the long arm of the X chromosome, band q27.(23),(24),(25),(26),(27) The full gene sequence is known and is ~31 kb in length and has 8 exons.(27) The mRNA is ~2.8 kb in length while the cDNA is 1386 bp (Table 1).(24),(27)
Loss-of-function mutations in the factor IX gene result in hemophilia B. The prevalence of this X-linked disorder is ~1 in 30,000 males, while females are only rarely affected. The five-fold difference in prevalence between hemophilia A and B is roughly equivalent to the difference in size of the coding portions of the factor VIII and IX genes. This is consistent with the fact that the chance of a given gene to be inactivated by mutation is largely dependent on its size. The clinical manifestation is almost identical to that for hemophilia A. Several mouse models of hemophilia B have been described and have proved very useful for gene therapy studies.(28),(29),(30)
The first mutation in the factor IX gene was described in 1983.(31) This mutation was a deletion that showed up in a screening using Southern blotting of genomic DNA. The first point mutation in factor IX, factor IX Chapel Hill, was discovered by protein sequencing, not by DNA analysis.(32) The pace of mutation detection changed dramatically after the introduction of PCR in 1987 and, at present, hundreds of different mutations are known in the factor IX gene.(33)
The various mutations associated with hemophilia B are registered in an international database, available on the internet (www.kcl.ac.uk/ip/petergreen/haemBdatabase.html). This hemophilia B mutation database focuses primarily on relatively small defects because, at most, gross gene alterations account for 5% of all hemophilia B cases. In this respect, hemophilia A and B differ significantly because, in the former disease, inversion events are the most common cause of severe disease. In 2004, the hemophilia B database was in its 13th edition. Some mutations appear to occur independently in patients from diverse geographic regions. These are mostly mutations at CpG, dinucleotides, the most common hot spot for point mutations. Overall, approximately one-half of the independent missense mutations found in different families with hemophilia B have been transitions within a CpG dinucleotide.
Of considerable interest are the 14 different mutations in the promoter region of the factor IX gene.(33) Two of these mutations are associated with conventional hemophilia B. The other promoter mutations are all associated with a certain form of hemophilia B (hemophilia B Leyden), arguably the most fascinating of all promoter mutations.(34),(35),(36) The unique feature of these variants is that individual's exhibit "recovery" from severe hemophilia with the onset of puberty. Thus, the disease is characterized by the absence of factor IX expression in childhood (factor IX levels less than 2%) and a gradual rise in factor IX levels after the onset of puberty. The increase in factor IX levels after puberty is probably related to increasing levels of testosterone.(37) Mutation detection in individuals with hemophilia B Leyden has revealed a heterogeneous group of mutations, all clustered at the 5' end of the gene.
The absence of factor IX expression during childhood in these individuals is theoretically straightforward to explain, inasmuch as all the mutations disrupt protein binding sites in the 5' flanking sequence but the pathophysiological basis for the gradual recovery after the onset of puberty has been more difficult to establish. Three sites appear to be involved, which are clustered on a 50-bp segment of DNA that includes the transcriptional start site. In the normal factor IX promoter, hepatocyte nuclear factor 4 (HNF-4) appears to bind to the two upstream elements, whereas the downstream element binds C/EBP (CCAAT-enhancer binding protein) or DBP (D-site binding protein) or both.(38),(39),(40),(41) The best explanation for the postpubertal recovery in hemophilia B Leyden lies in an androgen-responsive enhancer that partly overlaps one of the HNF-4 binding sites.(42),(43) Strong support for this hypothesis stems from the fact that mutations in this element lead to severe hemophilia B throughout life.
The relatively recent discovery of three factor IX mutations (Ala-10 to Val, Ala-10 to Thr; and Asn-9 to Lys) has revealed a unique type of genetic predisposition to bleeding during oral anticoagulation therapy attributable to increased warfarin sensitivity.(44),(45),(46) The Ala-10 and Asn-9 residues are within the factor IX propeptide region and are critical for interaction with λ-glutamyl carboxylase and thus λ-carboxylation; mutations at these sites impair this reaction. When receiving coumarins, patients with these variants showed a disproportionate decrease of factor IX activity approaching that of severe hemophilia B. As a consequence, these patients may suffer from bleeding at the very beginning of oral anticoagulation. After discontinuation of treatment, the factor IX levels return to normal.
Current Treatment Options
Intravenous protein replacement therapy is currently the mainstay of hemophilia care in the developed world. While this treatment regime has limitations, including cost, it is very effective and has helped thousands of patients. Early treatment, at the first onset of symptoms, limits both the amount of bleeding and the extent of the ensuing tissue damage. Replacement products for factor VIII or factor IX are obtained from either plasma-derived sources or they are produced as recombinant proteins from mammalian cell lines. Several products from either source are currently available. The reader is referred to the review by Kessler and Mariani(47) which gives an excellent overview of many of these products as well as general guidelines on using protein replacement therapy.
Unfortunately, ~20-30% of patients with FVIII deficiency and ~3-5% of patients with factor IX deficiency develop inhibitory antibodies to the plasma or recombinant-derived protein products. Low titer inhibitors can be overwhelmed with factor VIII or factor IX product but this is not feasible with high titer inhibitors. As a result, the research community sought to develop so-called "bypass strategies" which use other coagulation factors to provide hemostasis in these patients. The initial products included activated prothrombin complex concentrates (APCCs). While their precise mode of action remains somewhat obscure, it is thought that trace quantities of factor Xa, factor IXa and in particular factor VIIa within these products promote thrombin generation, thereby "bypassing" the need for the intrinsic pathway. Based on this idea, recombinant factor VIIa has been developed for the treatment of severe hemophilia complicated with inhibitors.(48) Recombinant factor VIIa was approved for use in the late 1990s and is now widely used to treat these patients. A drawback however is its short half-life (~2 hr), high dose requirement and significant cost. Another approach, at least for hemophilia A, is to use porcine factor VIII, which typically does not react with the existing inhibitory antibodies.(49),(50)
Future Treatment Options
As indicated above, the cost of treating hemophilia A or B patients with plasma or recombinant protein products is very expensive and the half lives of these proteins tend to be relatively short, necessitating frequent intravenous infusions. Over the past 20 years significant progress has been made by several research laboratories in understanding the biochemistry of factor VIII, factor IX and the hemostatic system in general. These studies have led to the development of novel approaches aimed at producing so called "third generation" recombinant protein products. Some of these approaches are discussed below. The reader however is referred to the following review articles for a more in-depth discussion.(17),(51),(52)
Protein production improvements: The expression of both factors VIII and IX in mammalian expression systems have limitations which principally relate to the complexity of their genes, posttranslational modifications and ability to be secreted in a functional form.
For factor VIII, several strategies have been applied to improve protein production. These include: 1) bioengineering factor VIII for more efficient expression of its mRNA by removing the B-domain or including introns in the cDNA sequence(53),(54) and 2) bioengineering factor VIII for more efficient secretion from the endoplasmic reticulum (ER) and Golgi. Strategies include changing the factor VIII primary sequence to reduce ER chaperone interactions(55),(56) and optimize interactions with trafficking proteins in the Golgi.(57)
Relative to factor VIII, the expression of factor IX is much more efficient in mammalian cells. However, several laboratories have made significant improvement in factor IX mRNA expression.(58),(59) One major limitation in the expression of factor IX, as well as other vitamin K-dependent, blood coagulation factors, is the efficiency of a critical posttranslational modification termed λ-carboxylation. This modification occurs in the N-terminal Gla domain and is critical for the proper function of factor IX. Depending on the level of protein expression and the cell type chosen, this modification can be sub-optimal. Our understanding of the mechanism of this reaction and the recent cloning and characterization of the genes and subsequent enzymes involved in this reaction has spurred new ideas on how to overcome limitations of this reaction in mammalian cells.(60)
Improvements in specific activity: Improving the functional properties of both factors VIII and IX has been explored as a way to potentially reduce the total dose of these proteins needed. Strategies for factor VIII include increasing its rate of activation through alteration of thrombin binding sites,(61) generating inactivation-resistant factor VIII by altering protease cleavage sites which are known to inactivate the molecule(62) and limit or eliminate A2 domain dissociation of factor VIII thereby increasing the apparent specific activity.(63),(64) For factor IX at least two reports have documented mutations in the catalytic domain that significantly increase the specific activity or potency of factor IXa.(65),(66)
Improvements in half-life: Notwithstanding improvements in pharmaceutical formulations, strategies to prolong the half-life of factor VIII include modifying its interaction with the low-density lipoprotein receptor-related protein (LRP), a hepatic clearance receptor with broad ligand specificity. The binding site on factor VIII for LRP has been partially characterized and thus mutations at this site in theory can improve the lifetime of factor VIII in vivo.(67)
Reduction in the immune response: As detailed above, inhibitory antibodies to factor VIII and factor IX develop in a significant number of patients. For factor VIII, the major targets for these antibodies have largely been identified (A2 and C2 domains). Based on the observation that these human inhibitory antibodies do not cross react with porcine factor VIII, hybrid human-porcine factor VIII derivatives have been produced. Several of these derivatives have been characterized and retain a high specific activity but do no cross-react with inhibitory antibodies directed against factor VIII.(68)
Gene Therapy for Hemophilia
As detailed above, hemophilia A and B are currently treated by the infusion of the missing clotting factor, mostly in response to bleeding episodes. While successful, there are numerous drawbacks to this type of approach, considering hemophilia is a lifelong disease and frequent infusions of these proteins are necessary. An attractive alternative would be to provide some level of the missing factor continuously in the circulation using a gene therapy strategy. The principles of gene therapy have been established in cell lines and several animal models over the past 15-20 years, and there are now numerous ongoing human gene therapy trials. In general, the approach involves genetic modification of a limited number of cells (typically with some viral vector) to produce a continuous amount of the deficient protein.
Hemophilia has a number of features that make it an attractive model for gene therapy. First, the therapeutic window is wide since even a modest elevation of factor VIII or IX (e.g., 2-4%) will result in a substantial improvement in clinical phenotype, and elevations of greater than 100% are unlikely to be harmful. Second, while clotting factors are normally synthesized in the liver, several studies have established that other cell types can synthesize biologically active proteins, thus expanding target options. Third, for both hemophilia A and B, there are excellent animal models (mice and dogs) facilitating pre-clinical studies before moving into human trials. Lastly, there are well established biological assays (ELISAs and clotting assays) to measure factor VIII and factor IX activity in plasma enabling rapid assessment of efficacy. Because of these and other factors, gene therapy for hemophilia A and B has moved forward from preclinical studies in animals to phase I trials in humans.(69) While some of the early results are encouraging in terms of safety, obstacles remain including immune reaction to the viral vectors.
Summary
Classical hemophilia is a relatively rare bleeding disorder resulting from a deficiency of either factor VIII or factor IX. These proteins assemble on an activated cellular surface to form the intrinsic Xase complex, one of two enzymes which activate factor X to factor Xa. Over the past two to three decades, the genes for these proteins and the genetic defects which cause hemophilia have been well described and range from single nucleotide changes to gross gene deletions. During this period, treatment options have significantly expanded and now include purified recombinant proteins which tend to be very safe compared to plasma concentrates used previously. In order to reduce cost, increase efficacy and reduce the likelihood of inhibitory antibodies against factor VIII or IX, the research community continues to bioengineer new recombinant products to circumvent some of these problems.