Course Authors
Frieda Wolf, M.D.
Release Date: 07/10/2006
Upon completion of this Cyberounds®, you should be able to:
Describe some milestones in the history of diabetic nephropathy
Discuss the pathologic cascade leading to diabetic nephropathy
List cellular mechanisms of injury due to hyperglycemia
Describe how podocyte injury is thought to contribute to nephropathy due to diabetes.
Dr. Wolf will discuss the unapproved use of ruboxistaurin mesylate in the prevention of diabetic nephropathy.
The discovery of insulin by Banting and Best in 1921 allowed individuals with Type 1 diabetes to live into their 30s. Ironically, the use of insulin enabled patients to live long enough to acquire organ damage such as nephropathy, retinopathy or cardiomyopathy.
Indeed, diabetic nephropathy has become the foremost reason for renal replacement therapy around the world today. Although genetic susceptibility and concomitant factors such as hypertension or hyperlipidemia can accelerate the progression of nephropathy, hyperglycemia is the main cause of damage at the cellular level.
In this Cyberounds®, we will summarize some milestones in the pathophysiology of diabetic nephropathy and review pathways that converge on the overproduction of superoxide by the mitochondrial electron transport chain. The overproduction of superoxide is thought to be the unifying mechanistic step in diabetic tissue damage at the cellular level. We will address Type 1 and Type 2 diabetic nephropathy interchangeably, as hyperglycemia-induced tissue damage is common to both. Finally, we will see that the rate of diabetic nephropathy has started to decline in the United States, and this may reflect ongoing efforts to control blood pressure and glycemia since the DCCT and UKPDS trials have pointed in that direction.
A Brief History of Diabetic Nephropathy
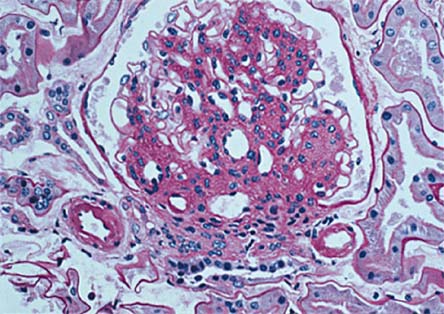
The first description of diabetes-induced injury in the kidneys, "glycogen nephrosis," was recounted by Woredekal and Friedman (2006).(1) Even though glycogen deposition in renal tubular cells, cardiac muscle and pancreatic beta cells was described in diabetic individuals(2) in the late 1870s by Armanni and early 1880s by Ebstein, it was not until 1936, when Kimmelstiel and Wilson(3) noted hyaline thickening of the intercapillary connective tissue with formation of nodules (see Figure 1), that diabetic nephropathy, the syndrome of nephrotic edema, proteinuria and hypertension, became synonymous with Kimmelstiel-Wilson Disease.

Figure 1. Diffuse and Nodular Diabetic Sclerosis.
The clinical picture of diabetic nephropathy became more detailed when, in 1969, Keen(4) and colleagues reported abnormal increase in urinary albumin, and the term microalbuminuria was born. Microalbuminuria was later linked by Mogensen(5)and colleagues in the 1970s to Type I diabetes, nephromegaly and glomerular hyperfiltration. Subsequently, in 1984, Mauer and colleagues concluded that "the critical lesion of diabetic nephropathy, which ultimately leads to organ failure, is the expansion of the glomerular mesangium."(6) However, researchers did learn that after a decade of euglycemia these lesions are reversible.(7) What seems, therefore, to be clearly established is that the progression of glomerulopathy in diabetic individuals is a result of hyperglycemia.
Pathophysiology of Hyperglycemia-induced Damage
The pathway between hyperglycemia and end organ damage has been under investigation since the 1960s. Diabetic nephropathy is first manifested by glomerular hyperfiltration, which leads to microalbuminuria and overt proteinuria. As glomerular filtration rate (GFR) deteriorates, chronic renal failure and its sequelae follow.(8) As end stage renal disease ensues, renal replacement therapy with dialysis or transplantation is required. Pathologically, lesions of diabetic nephropathy manifest with expansion of extracellular matrix in the mesangium, which is associated with declining GFR, as less glomerular surface area is available for filtration.(9) Some common molecular mechanisms are thought to operate simultaneously to cause these lesions.
Hyperglycemia-induced glomerular hyperfiltration is the result of the dilation of the afferent glomerular arteriole to a greater extent than the efferent arteriole. This unequal dilation raises the intraglomerular hydrostatic pressure causing an increase in the passage of ultrafiltrate through the glomerular filtration apparatus. These hemodynamic changes are thought to be mediated by increased production of vasodilatory prostanoids(10) and nitric oxide.(11)
Increased filtration is soon followed by microalbuminuria, the manifestation of the damaged filtration apparatus of the basement membrane. This phenomenon has been attributed to changes in the synthesis and catabolism of various glomerular basement membrane macromolecules, such as collagen and proteoglycans, which together provoke thickening of the basement membrane (GBM). Another possible explanation for the increased glomerular permeability is the rise in renal vascular endothelium growth factor (VEGF)(12) levels that are observed in preclinical models of diabetes, since VEGF is both an angiogenic and a permeability factor.
Diabetic nephropathy has been associated with mesangial expansion and mesangial cell alterations since the first description by Kimelstiel and Wilson. Mesangial expansion is associated with a thickening basement membrane, which allows less capillary surface area to be available for filtration. Mesangial expansion appears to be dependent on the action of growth factors, especially transforming growth factor-beta(13) and possibly connective tissue growth factor.(14) This increase in TGF-β results in increased type I and IV collagens, as well as decreased proteoglycans, producing a thickened basement membrane.
Cell culture studies have demonstrated that increases in glucose concentrations can induce TGF-β expression and, in concert with vasoconstrictor or vasodilator imbalance,(15) stimulate extracellular matrix growth in a manner similar to that observed in diabetic nephropathy in vivo. Hyperglycemia as well as poor blood pressure control can accelerate the progression of diabetic nephropathy.(16) Both processes contribute to the microvascular damage that leads to nephropathy.
Hemodynamic alterations, endothelial dysfunction, inflammatory cell activation and fluctuations in the expressions of vascular and neurotrophic factors all combine to induce local tissue-specific pathological changes.(17) For example, thickened basement membrane has pathologic consequences for glomerular function but not for muscle tissue. Tubulointerstitial fibrosis then contributes to renal failure and it is further exacerbated by proteinuria, which in itself is injurious to the interstitium and worsens fibrosis.(18)
Though mesangial expansion and increasing GBM thickness correlate with membrane porosity and proteinuria, they do not explain proteinuria. The clinical syndrome of diabetic nephropathy is the nephrotic syndrome, proteinuria being its main manifestation. As we'll see later, podocyte biology may provide an explanation. [Since GBM thickening and mesangial expansion are chronic and are modified by systemic factors such as hypertension, poor blood pressure control can accelerate the progression of nephropathy.] Furthermore, genetics may play a role in determining which organ will incur overt damage in an individual(19) and why end-stage renal disease (ESRD) disproportionately affects racial minorities.
Cellular Mechanisms of Injury
Researchers have elucidated several mechanisms of injury at the cellular level:
- The aldose reductase theory seeks to explain the development of cataracts: hyperglycemia increases conversion of glucose to sorbitol, which contributes to cataract formation since the lens has a high level of aldose reductase. This contributes to the consumption of NADPH, a cofactor for both aldose reductase and for glutathione, increasing oxidant stress.
- AGE theory: advanced glycation endproducts are proteins that become irreversibly glycated by sugars in a nonenzymatic process known as the "browning" reaction. AGEs can alter structural proteins such as collagen and cellular function. Cellular function is changed because AGEs bind to cellular receptors known as RAGE (receptors for AGE). RAGEs are transmembrane proteins which activate a series of cellular signaling events such as mitogen-activated-protein (MAP) kinase or protein kinase C (PKC). These are signals for gene transcription to occur. Unfortunately, these genes produce inflammatory cytokines and growth factors. Aminoguanidine, an inhibitor of AGE formation, has disappointed in clinical trials, though its lack of efficacy may be the result of addressing a single molecular pathway to glucotoxin formation.
- Hexosamine pathway: normally glucose is metabolized in the glycolytic pathway. In hyperglycemia some glucose in diverted to the GFAT (glutamine:fructose-6 phosphate amidotransferase) pathway. This pathway activates transcription of genes such as TGF-β
- Protein kinase C: these intracellular enzymes govern phosphorylation of specific sets of proteins, such as tyrosine, serine or threonine, by transfer of phosphates from ATP. Elevated glucose increases levels of diacyl glycerol (DAG) through the glycerol-3-phosphate pathway. Elevated DAG activates PKC. Activation of PKC, in turn, causes vascular damage that includes nitric oxide dysregulation, increased permeability and increased leukocyte adhesion. Growth factors such as vascular endothelium growth factor (VEGF) and transforming growth factor beta (TGF-β)(23) are also increased by PKC. PKC also activates the intracellular MAP pathway.(24) When PKC is activated, gene expression is altered, decreasing expression of genes that are protective, and increasing expression of genes that are harmful. There are several isoforms of PKC and in glomeruli the α, β, γ, and ε isoforms are activated. Ruboxistaurin mesylate, a PKC inhibitor with affinity for the β isoforms, blocks endothelial cell abnormalities and can prevent/reverse diabetic nephropathy in animals with diabetes.(25) Currently in clinical trials, ruboxistaurin mesylate appears to address the PKC mechanism by which glucose is toxic to cells -- activation of MAP kinase. Ruboxistaurin mesylate specifically inhibits the β isoform of PKC and, therefore, does not affect other intracellular signaling pathways which rely on the PKC kinase system.
There seem to be overlapping pathways of tissue injury by hyperglycemia. These pathways intertwine to amplify vascular and tissue damage, and converge on a single hyperglycemia-induced process, the overproduction of superoxide by the mitochondrial electron transport chain.(26) This has been termed the unifying mechanism by Michael Brownlee and in its center is the free radical superoxide. The latter is a free radical that is generated by increasing oxidative phosphorylation in the mitochondria. Free radicals can damage cellular proteins and they also decrease nitric oxide levels, further damaging cellular proteins. Hyperglycemia induces superoxide production, which in turn inhibits GAPDH (glyceraldehyde-3-phosphate dehydrogenase). This causes an increase in molecules "upstream" from this enzyme. Increase in glyceraldehyde-3 phosphate activates the AGE pathway and the PKC pathway. Increases in fructose-6-phosphate drives GFAT and the hexosamine pathway. Finally, increase in glucose activates aldose reductase, consuming NADPH.
Diabetic individuals appear to have increased levels of oxidative stress as evidenced by elevated LDL.(27) Theoretically, the use of an antioxidant such as lipoic acid(28) or high-dose vitamin E(29) may improve some hemodynamic changes in the kidney but clinical proof is still lacking. Furthermore, addressing only one molecular mechanism, as in the inhibition of AGEs above, may be insufficient to prevent or reverse diabetic nephropathy.
Podocyte Injury
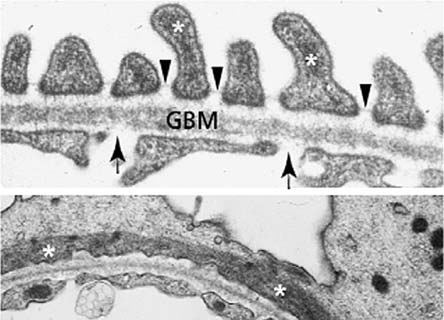
Although renal tissue nephropathy has focused on mesangial cell injury, visceral epithethial cells (podocytes) may contribute significantly to nephropathy.(30) The glomerular filtration barrier is composed of glomerular capillary endothelium, the basement membrane and the podocytes. The foot processes of podocytes interdigitate and form areas of porous membrane called the slit diaphragm (Figures 2 and 3). The integrity of the slit diaphragm is the primary determinant of the selectivity of the filtration barrier.
Figure 2. Electron Microscopy (EM) of Glomerular Basement Membrane (GBM).
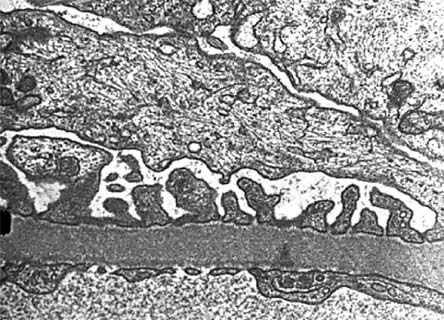
Figure 3. Slit Diaphragms, Glomerular Basement Membrane (EM)ine.
Nephrin is a large transmembrane protein that forms the slit diaphragm and via proteins such as podocin and CD2-AP (CD2 associated protein) connects the slit diaphragm with the cytoskeleton of the podocyte (Figure 4). Signaling through an intact slit diaphragm via nephrin will prevent podocyte apoptosis.
Figure 4. Figure HeadCartoon of Zipper-like Structure of the Filtration Slit of the GBM.
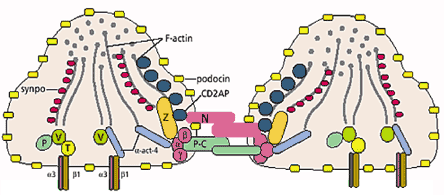
Glomerular Basement Membrane (GBM)
Source: Dr. Eli A. Friedman
Podocytes do not normally divide. In diabetic nephropathy, podocytes undergo widening of their foot processes, as well as reduction in number and density. This has been termed podocytopenia.(31) Podocytes may be found in the urine of diabetic individuals (podocyturia) with microalbuminuria or overt proteinuria,(32) as compared with normoalbuminuric patients and healthy controls. The TGF-β system in the podocyte also promotes podocyturia. Podocytopenia may stimulate synechiae formation, the first step in glumerulosclerosis, by increasing contact of the 'denuded' GBM with Bowman's capsule.
The narrowing of the slit diaphragm may impede the filtration of water thus reducing GFR. When the amount of protein relative to water in the tubule exceeds the reabsorptive capacity of the tubules for proteins, 'spillover,' manifested as proteinuria, occurs. This theory may explain why in diabetic nephropathy there is increasing proteinuria despite declining GFR. Proteinuria itself induces tubulointerstitial fibrosis(33) and in concert with glomerulosclerosis leads to chronic renal insufficiency. Angiotensin II may play a role in podocyte injury. It appears that use of an ACE inhibitor or AT1 (angiotensin 1) receptor blocker attenuates the reduction of nephrin expression in diabetic rat models.(34) This may be the mechanism for the antiproteinuric effect of these drugs.
The structure of the GBM may also change in diabetic nephropathy. There is decreased synthesis of heparin sulfate proteoglycans because of increased generation of reactive oxygen species. Podocytes are the main source of proteoglycans, especially agrin. High glucose and angiotensin II suppress production of agrin, the primary proteoglycan.(35) Furthermore, podocytes are the main source of VEGF in the glomerulus and its expression is stimulated by high glucose, by TGF-β and by angiotensin II via AT1 and AT2 receptors. Higher concentration of VEGF stimulates production of collagen IV in the basement membrane, thereby contributing to its increased thickness and altered permselectivity of the GBM. This, in turn, suppresses nephrin expression, favoring apoptosis of pododytes, with foot process effacement.
Nephropathic Damage
The damage of diabetic nephropathy has several faces. There is ongoing vascular damage concomitant to interstitial tissue injury which affects hemodynamics and stimulates fibrosis and is termed microvascular damage. There is also ongoing macrovascular damage, as increased free fatty acids are released from adipocytes into endothelial cells and cause an overproduction of reactive oxygen species. These, in turn, activate the same damaging pathways discussed above, AGEs, PKC and hexosamine.(38) It has been difficult to reach a unifying theory regarding mechanisms of damage and even more difficult to design therapeutic approaches. Some, however, have tried to suggest common mechanisms, which, taken together, result in pathology.
New insights into cellular mechanisms of kidney damage produced by excess protein have been offered by Tuttle,(37) who recently demonstrated that a profibrotic injury response occurred in mesangial cells exposed to amino acids, with or without high glucose, by formation of AGE (advanced glycation end-products of nonenzymatic protein glycosylation), oxidative stress and activation of the PKC using the MAP kinase-ERK 1,2 (extracellular signal regulated kinase) signal pathway.
The mesangial cell is involved in glomerular hemodynamics, as well as fibrosis and matrix production in the interstitium. It may also be involved in the excess formation of AGEs by increasing availability of free amino groups. When mesangial cells were cultured with a high level of amino acids, similar to the profile observed in the circulation after a protein meal, they responded with profibrotic injury. Interestingly, their response was remarkably similar to that observed when they were cultured in high glucose. Mesangial cell activity is modulated by protein phosphatases, which remove the phosphate groups. Hyperglycemia and AGEs increase reactive oxygen species, activating protein kinases such as NFκB, protein kinase C and TGF-β.(38) These kinases cause increased extracellular matrix and, secondarily, renal hypertrophy. Mechanisms leading to glucose- and amino acid-mediated cellular responses may be shared.
A Common Molecular Mechanism of Hyperglycemic Damage?
These recent observations suggest that there is a common molecular mechanism for hyperglycemia-induced damage(39) in which mitochondria are required for initiation of superoxide production. It appears that overproduction of the reactive free radical superoxide is the genesis of a vicious cycle of oxidative stress that leads to accelerated atherosclerosis and the microvascular complications of diabetes. Production of superoxide then amplifies the damaging effects of hyperglycemia.
Wider fluctuations in glucose in the postprandial state may trigger greater oxidative stress than chronic sustained hyperglycemia.(40) This may explain findings in the DCCT trial which showed that diabetics with similar HbA1c values but greater blood glucose fluctuations were more likely to have retinopathy; it is not the time-averaged glycemia but rather the magnitude of fluctuation which generates a bigger free-radical load and the consequent initiation of atherosclerosis. Prostacyclin synthase is an antiatherogenic endothelial cell enzyme that prevents atherosclerotic plaques through actions on endothelial cells, monocytes, macrophages and vascular smooth muscle cells. Its levels are remarkably decreased in oxidative stress by free radicals.(41)
Future Trends
Much progress has been achieved in the fight against diabetic nephropathy. According to NHANES, the prevalence of diabetes in the United States is greater than ever,(*) estimated at 6.5% of the 2002 US population of diagnosed and 2.8% of the US population of undiagnosed individuals with diabetes, totaling 19.3 million people. Yet, as reported by the CDC,(43) the incidence of ESRD from diabetes is declining, with age-adjusted incidence decreasing by 21% from 293/100,000 in 1997 to 232/100,000 in 2002 (Figure 5).
Figure 5. Age-adjusted Incidence of Diabetic Nephropathy.
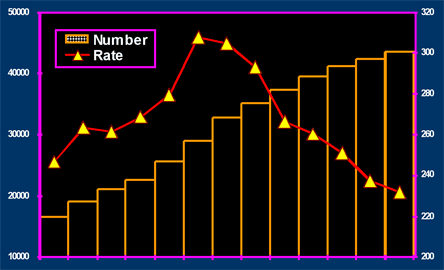
Source: Dr. Eli A. Friedman
USRDS: 2006
Conclusion
Existing strategies for treating nephropathy include:
- strict glycemic control with minimal glycemic fluctuations
- optimal blood pressure control
- blockade of the renin-angiotensin system.
Each of these approaches helped to achieve the observed decline in incidence of diabetic nephropathy. Additionally, potential novel strategies including PKC inhibitors, now in clinical trials, inhibitors of oxidative stress and fibrosis, and perhaps even AGE inhibitors, if matched with other clinical initiatives, may further reduce incidence. It is hoped that the tight control of hyperglycemia, the restoration of euglycemia at the cellular level and these potential novel molecular targets of therapy may one day lead to the eradication of diabetic nephropathy.
The hope is that these strategies, in combination with novel molecular targets of therapy, may lead to the eradication of diabetic nephropathy.