Course Authors
Harold R. Collard, M.D., and Talmadge E. King, Jr., M.D.
Release Date: 06/07/2006
Upon completion of this Cyberounds®, you should be able to:
Discuss the current approach to the diagnosis of idiopathic pulmonary fibrosis
Describe the natural history of idiopathic pulmonary fibrosis and how to approach prognosis
Describe the data behind potential therapies for idiopathic pulmonary fibrosis and establish an approach to management that is evidence-based.
Drs. Collard and King will discuss the unapproved and investigational use of cyclophosphamide, azathioprine, acetylcysteine, interferon gamma 1b, bosentan, pirfenidone and warfarin for the treatment of idiopathic pulmonary fibrosis.
|
Case Report
A 65-year-old man presents with shortness of breath and cough. Over the last six months he has had increasing breathlessness with exertion. He has a dry cough that is worse with exertion. He is a former smoker (30 pack-years) but quit 10 years ago. His medical history is remarkable for gastroesophageal reflux. He is a retired teacher and has no identifiable occupational or environmental exposures. There is no family history of lung disease. On physical examination, he has inspiratory crackles. |

Work-Up of Diffuse Parenchymal Lung Disease
Patients with diffuse parenchymal lung disease (DPLD) commonly come to clinical attention with the onset of progressive shortness of breath with exertion (i.e., dyspnea), persistent cough, or the identification of interstitial opacities on chest x-ray. In patients with common forms of DPLD such as idiopathic pulmonary fibrosis (IPF) and sarcoidosis, the symptoms and signs are chronic (i.e., months to years). However, the symptoms may be acute (i.e., days to weeks) or subacute (i.e., weeks to months) in some patients with DPLD.
The initial evaluation of suspected DPLD should include a complete history and physical examination. The initial laboratory evaluation should include biochemical tests to evaluate liver and renal function and also hematologic tests to check for anemia, polycythemia or leukocytosis. Serologic studies should be obtained if clinically indicated by features suggestive of an autoimmune disease or vasculitis: these tests should include sedimentation rate, antinuclear antibodies, rheumatoid factor, antineutrophil cytoplasmic antibodies, proteinase 3 and myeloperoxidase levels, and antibasement membrane antibody. A recent chest x-ray should be obtained and it is important to review all old chest x-rays to assess the rate of change in disease activity.
High-resolution computed tomography (HRCT) is better than the chest x-ray for early detection and confirmation of suspected DPLD. HRCT also allows better assessment of coexisting disease (e.g., emphysema, mediastinal adenopathy or cancer). Complete lung function testing (i.e., spirometry, lung volumes and diffusing capacity) and resting room air arterial blood gases should be obtained. Most causes of DPLD will be identified by this approach.
The patient's chest x-ray showed bilateral bibasilar reticular opacities with a normal sized heart. His forced vital capacity (FVC) was only moderately reduced (65% of predicted), his FEV1/FVC ratio was 85%, his total lung capacity (TLC) was 62% of predicted and his diffusion capacity for carbon monoxide (DLCO) was 45% of predicted. His resting room air oxygen saturation was 92%. On walking in the hall, he was noted to desaturate to 85%. An HRCT was obtained and revealed a pattern of diffuse, bilateral, subpleural reticular opacities, traction bronchiectasis and patchy areas of cystic "honeycomb" change.
Diagnosing DPLD
High-resolution CT scanning has revolutionized the diagnostic approach to DPLD. In many instances, the proper clinical history and HRCT appearance provide a definitive diagnosis, obviating the need for a surgical lung biopsy. In this case, the clinical presentation, the restrictive pattern on pulmonary function testing and the HRCT scan meet criteria for the diagnosis of IPF (Table 1).
Table 1. Clinical Criteria for the Diagnosis of IPF.
Major Criteria
|
* Modified from reference 1. Non-diagnostic bronchoscopy is now generally dropped from the criteria.
Diagnosis of IPF
IPF is a disease of older age, with the mean age of diagnosis around 60 to 65 years. Its estimated incidence is around 30 cases per 100,000 people. Men are more commonly affected than women and the overwhelming majority of patients are former smokers. The history and physical are nonspecific, although clubbing is more common in IPF than other forms of diffuse lung disease and should suggest the diagnosis.
Several studies have shown that the clinical/radiographic diagnosis of IPF is highly specific (>90%).(1),(2) The accuracy of the clinical diagnosis is largely driven by the high-resolution computed tomography (HRCT) scan appearance (Figure 1).(3)
Figure 1. High Resolution Computerized Scan of Chest in IPF.
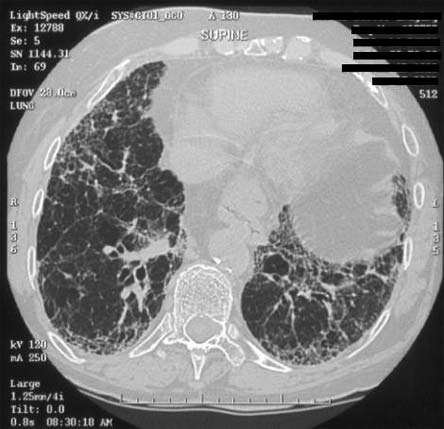
High resolution computed tomography (HRCT) scanning of the chest is diagnostic in some patients with IPF. The presence of bi-basilar, peripheral reticular abnormality, traction bronchiectasis, honeycomb cysts and minimal ground glass are classic findings.
Approximately half of patients with IPF will have diagnostic HRCT appearances and can be confidently diagnosed without undergoing surgical lung biopsy. Importantly, there remain many cases of IPF with atypical HRCT scans; these still require a surgical lung biopsy for accurate diagnosis.
Bronchoscopy has historically been used to evaluate patients with suspected IPF but its diagnostic utility has recently been questioned. There are no findings on bronchoalveolar lavage specific to IPF and transbronchial lung biopsy does not provide adequate tissue for a histopathological diagnosis. Its primary role remains ruling out alternative diagnoses such as infection or malignancy. Figure 2 shows the authors' approach to the diagnosis of IPF.
Figure 2. Diagnostic Algorithm for Suspected IPF.
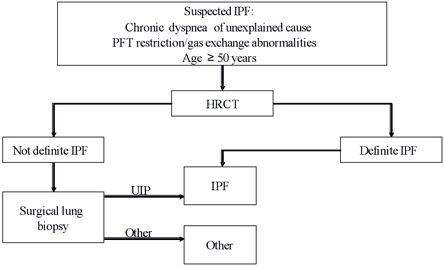
All patients suspected of having IPF should undergo HRCT scanning. If the HRCT is diagnostic, a surgical lung biopsy can be avoided. In all other patients, surgical lung biopsy should be considered.
Histopathologic Features of IPF
The definition of Idiopathic Pulmonary Fibrosis (IPF) has changed over the last twenty years from a general description for diffuse lung disease of unknown cause to a specific clinicopathologic entity defined by the histopathological presence of usual interstitial pneumonia pattern (Figure 3).(4)
Figure 3. Lung Histopathology in IPF -- UIP Pattern.
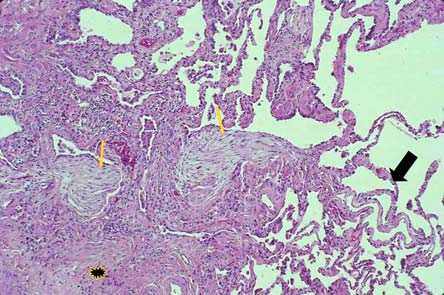
Usual interstitial pneumonia pattern is characterized by patchy subpleural dense fibrosis (star), focal areas of early collagen deposition termed "fibroblast foci"(narrow arrows), adjacent to areas of normal appearing lung (large arrow) and a general absence of extensive lymphoplasmacytic inflammation.
This change was in part motivated by the finding that patients with usual interstitial pneumonia pattern had a distinctly worse prognosis than those with other histopathological patterns.(5),(6),(7)
Usual interstitial pneumonia pattern is characterized by:
- "temporal heterogeneity" of lung fibrosis (meaning that areas of end stage fibrosis are adjacent to areas of early fibrosis and areas of normal lung),
- localized areas of proliferating fibroblasts and collagen deposition termed fibroblast foci,
- microscopic cystic spaces lined by bronchiolar epithelium and
- minimal lymphoplasmacytic inflammation which, if present, is generally confined to areas of advanced fibrosis.
Prognosis in IPF
Predicting survival in IPF remains a challenge for clinicians. While the average survival is between two and three years from the time of diagnosis, there is great variability in individual patients' courses. Numerous papers have been published on clinical and physiological predictors of survival such as age, smoking status and baseline pulmonary function values with largely mixed results. Recently, several authors have reported change in FVC over 6 or 12 months to be an important predictor of survival.(8),(9) Change in dyspnea score, alveolar-arterial oxygen gradient and diffusing capacity for carbon monoxide.
(DLCO) have also been reported to be predictive of survival, although less reliably.(8),(10) In patients with fibrotic interstitial pneumonia, changes in physiology may, in fact, trump histopathology as the more powerful predictor of outcome.(10)
Other predictors have been recently identified, although they are less well validated than FVC. The presence of desaturation on 6-minute walk test (defined as saturations <88%) and the presence of pulmonary hypertension on right heart catheterization have been reported as predictors of survival.(11),(12) Unfortunately, echocardiography is inaccurate in estimating the presence of pulmonary hypertension in patients with IPF. The HRCT scan appearance also adds important prognostic information, as patients with IPF and atypical (i.e., non-diagnostic) HRCT images survive significantly longer than those with more typical features.(3)
The patient wants to know what treatment options are available to improve his likelihood of survival and maintain or improve his quality of life.
Treatment
There remain no proven therapies for IPF, although several preliminary studies show promise (Table 2).
Table 2. Selected Studies of Therapy in IPF.
| Stuay | Drug | Design | Result |
|---|---|---|---|
| Douglas et al.(17) | Prednisone | Retrospective comparison of prednisone and no therapy | No difference in survival (p=0.72) |
| Collard et al.(8) | Prednisone and cyclophosphamide | Retrospective comparison of prednisone/ cyclophosphamide and no therapy | No difference in survival (p=0.58) |
| Demedts et al.(18) | Prednisone, azathioprine and acetylcysteine | Randomized double-blinded trial of prednisone/azathioprine/acetylcysteine vs prednisone/azathioprine alone | Slower decline in FVC over 12 months with acetylcysteine (1% vs. 5% decline, p=0.02) |
| Raghu et al.(20) | Interferon gamma 1b | Randomized double-blinded placebo-controlled trial of interferon gamma 1b | No difference in "progression-free survival" defined >10% decrease in percent predicted FVC, >5% increase in Aa gradient, or death (p=0.5) |
| Azuma et al.(21) | Pirfenidone | Randomized,double-blinded, placebo-controlled trial of pirfenidone | No difference in "change in lowest oxygen saturation on 6 minute walk test" over 9 months; slower decline in FVC over 9 months with pirfenidone (0.03L vs. 0.13L, p=0.04) |
| Kubo et al.(22) | Anticoagulation | Randomized, open-label, placebo-controlled trial of anticoagulation | Improved mortality with anticoagulation (Hazard ratio (no anticoagulation) 2.9 (95%CI 1.0, 8.0, p=0.04) |
Despite significant insight into the pathobiology of fibrosis and substantial investments of time and money in treatment trials, there remain no definitive studies showing a treatment effect in IPF when compared to no therapy. This review will discuss the following therapies:
- cortocisteroid plus azathioprine or cyclophosphamide,
- acetylsysteine,
- interferon gamma 1b,
- pirfenidone and
- anticoagulation.
Prednisone Plus Aazathioprine or Cyclophosphamide
Combination therapy with corticosteroids and cytotoxic agents has been the standard of care in IPF for many years. Prospective but uncontrolled studies have shown up to a third of patients "responding" to this therapy, generally by improving their breathlessness or pulmonary function.(8),(13),(14) Retrospective studies have failed to show a mortality benefit for corticosteroids and cytotoxic agents when compared to untreated patients.(16),(17) Unfortunately, no randomized, placebo controlled trial has ever been performed and the role of this regimen in the treatment of IPF remains unclear.
Acetylsysteine
Acetylcysteine is a precursor to glutathione, a potent antioxidant on the lung, and it has been hypothesized that therapy with acetylcysteine may be effective in the treatment of IPF. A large trial of "standard therapy" i.e., prednisone plus azathioprine) compared to combination prednisone, azathioprine and acetylcysteine showed a clinically and statistically significant difference of 4% in the primary outcome measure of change in forced vital capacity (FVC) over 12 months.(18) This endpoint has been shown to be a reliable surrogate for survival time.(19) There are no placebo controlled data of acetylcysteine. Whether or not acetylcysteine alone or combination therapy with prednisone, azathioprine and acetylcysteine is better than no therapy remains unanswered.
Interferon gamma 1b
Interferon gamma 1b has been shown to attenuate tumor growth factor (TGF) beta-mediated fibrosis. A large, placebo controlled trial of interferon gamma 1b in IPF failed to show a difference in the primary outcome of progression-free survival (defined as a change in pulmonary function, gas exchange or death) over a median follow up period of one year.(20) Subgroup analysis suggested that there may be a mortality difference in patients with preserved pulmonary function, and a larger, longer study of interferon gamma 1b is currently underway, with results expected in 2008.
Pirfenidone
Pirfenidone is an antifibrotic agent that appears to attenuate TGF beta-mediated fibrosis. A recent phase 2 study of pirfenidone compared to placebo showed a slower decline in FVC over 9 months in the pirfenidone group.(21) This has prompted a larger, more definitive phase 3 trial that is currently enrolling patients.
Anticoagulation
Coagulation has long been implicated in inflammation and fibrosis and recently inhibition of thrombin has been shown to prevent fibrosis in animal models. A small study of anticoagulation (warfarin and low molecular weight heparin) in patients with IPF showed that anticoagulation improved survival when compared to no therapy.(22) These data are encouraging but need to be confirmed in larger, better-designed trials.
Other Agents
There are many other agents currently under investigation in phase 1 and 2 studies. A phase 2 trial of bosentan [an endothelin receptor (ET-1) antagonist] vs. placebo has been completed and preliminary results show no difference in the primary endpoint of 6-minute walk distance but possible effects on mortality in selected patients. Etanercept (a tumor necrosis factor receptor antagonist) has also been studied in a phase 2 trial, the results of which remain unpublished. Other agents such as imatinib mesylate, sirolimus, sildenafil, connective tissue growth factor inhibitors, thrombin inhibitors and anti-TGF beta agents are in the early stages of clinical investigation.
The patient decides to forgo treatment for now given the lack of evidence for efficacy. You arrange to follow the patient's pulmonary function (in particular FVC) over the next six months to better inform his prognosis. Two months later, you receive a call that the patient has had an abrupt worsening of their condition and has just been admitted to the hospital.
Patients with IPF may develop acute worsening of their disease without evidence of a secondary identifiable etiology such as infection or pulmonary embolism. This idiopathic acute worsening has been termed acute exacerbation of IPF.(23),(24) Acute exacerbation is generally defined by the following criteria:
- subjective worsening in dyspnea over 30 days or less,
- new ground glass abnormality on chest radiography,
- worsening oxygenation and
- absence of infection, pulmonary embolism, cardiac event or pneumothorax.
When biopsied, these patients generally demonstrate diffuse alveolar damage, the same histopathologic pattern seen in acute interstitial pneumonia and acute respiratory distress syndrome (Figure 4).
Figure 4. Histopathology in Acute Exacerbation of IPF -- DAD Pattern.
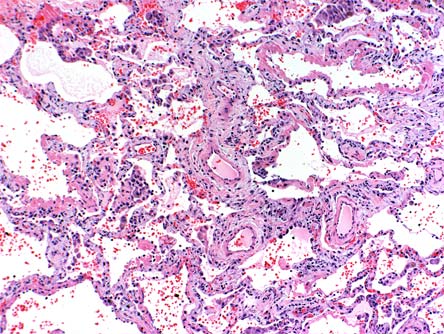
Diffuse alveolar damage pattern demonstrates alveolar wall thickening, collagen, type II pneumocyte hyperplasia and hyaline membranes. This pattern of acute lung injury is seen in IPF patients with acute exacerbation of their disease and is identical to that seen in acute interstitial pneumonia and the acute respiratory distress syndrome.
The incidence of acute exacerbations of IPF is unknown with estimates ranging from 10% to over 50% annually.(21),(22) Acute exacerbations appear to be highly morbid and a common cause of death in patients with IPF.(25) Data from recent clinical trials have suggested that the natural history of IPF may be less that of a slow, steady decline and more that of periods of relative stability punctuated by acute exacerbation (Figure 5).
Figure 5. Evolving Concept of the Natural History of IPF.
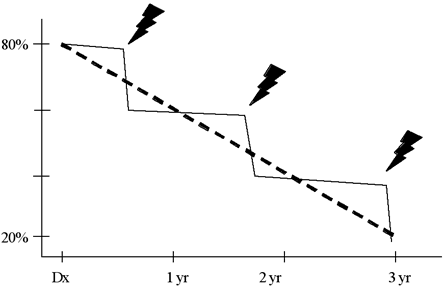
The traditional paradigm suggests IPF causes a gradual decline in pulmonary function over time (dotted line). An alternative paradigm describes a more episodic course (solid line), with long periods of overall stability punctuated by acute worsening (lightning bolts) that are associated with high morbidity and mortality.
Treatment of acute exacerbations of IPF generally involves high-dose corticosteroid and antibiotics but there are no good data to support this approach. It appears that pirfenidone and anticoagulation may both be effective in reducing the incidence and morbidity of acute exacerbation but additional data are needed.(21),(22)
The patient recovered and was discharged from the hospital but he is now more hypoxemic and symptomatic with dressing and undressing. He returns to clinic for follow-up and asks about other treatment options. He also tells you that his hospital doctor told him during his recent hospitalization that he was on the verge of needing mechanical ventilation and the patient wants to know your opinion on whether this would be a reasonable intervention should he worsen again in the future.
Non-pharmacological approaches to therapy are a critical component of patient management in IPF. While no data specific to IPF exist to prove efficacy, it is generally believed that the treatment of hypoxemia (oxygen saturations below 88%) with supplemental oxygen and involvement in pulmonary rehabilitation are beneficial. It is the authors' opinion that these should be prescribed in all eligible patients.
Lung transplantation has become an important therapy for IPF in eligible patients with advanced disease, as it has been shown to improve survival.(26) In general, patients with advanced IPF, age 65 or less, with limited comorbidities and good social support should be referred to a lung transplant center for evaluation. The definition of advanced IPF is generally based on severely decreased FVC (<50% predicted) or DLCO (<40% predicted) values. Several studies have suggested that patients with a DLCO value of 39% predicted or less should be referred for transplantation evaluation.(27),(28) This recommendation was based on an average waiting time to transplantation of approximately two years. Although newly implemented prioritization schema may shorten this wait time, it is still prudent to promptly refer all potentially eligible patients with advanced IPF for evaluation, as the risk of acute exacerbation and death appears substantial in all IPF patients.(19)
Mechanical ventilation in patients with IPF has a very poor prognosis.(29),(30) One study of 38 patients admitted to the ICU found no impact of mechanical ventilation on survival and reported a 2-month survival rate of 8%.(30) These data highlight the importance of end-of-life care discussions with patients with IPF.