Course Authors
Sam Engel, M.D.
Release Date: 07/20/2005
Upon completion of this Cyberounds®, you should be able to:
Discuss the regulation of glucose homeostasis by incretins
Describe the abnormalities in incretin levels in Type 2 diabetes
Discuss the therapeutic approaches to restoring the incretin effect in Type 2 diabetes.
Dr. Engel will discuss the unlabeled use of liraglutide and DPP-IV inhibitors for Type 2 diabetes.
The understanding of the pathophysiological mechanisms that underlie the development of Type 2 diabetes has evolved over the past two decades. Our view has shifted -- from a primary focus on insulin resistance to a more comprehensive approach that recognizes the dual defects of insulin resistance coupled with a relative deficiency in insulin secretory capacity.
While insulin resistance is considered to be the primary defect of Type 2 diabetes, and is typically the result of obesity and/or a genetic predisposition, most individuals with insulin resistance are able to maintain normal blood glucose levels by compensatory hypersecretion of insulin. It is only when this compensation does not occur that disorders of glucose tolerance arise.
The traditional view of the beta cell defect in Type 2 diabetes has been one of a "quantitative" defect, wherein the pancreas gradually loses the ability to hypersecrete insulin. This progressive failure in beta cell capacity has been attributed to both beta cell "exhaustion" as well as loss of beta cell mass. However, more recently, the appreciation of "qualitative" defects in beta cell function has emerged, and has resulted in the development of new therapeutic strategies for Type 2 diabetes.
Defects in Beta Cell Function
The islets of Langerhans represent only about 2% of the pancreas by weight, with the majority of the islet being comprised of beta cells. The principle function of the beta cell is the secretion of insulin in response to nutrients, hormones and neuronal stimuli.(1) Glucose, the primary regulator of insulin secretion, induces the secretion of insulin through a series of signaling pathways that have been elucidated over the past two decades.
Glucose enters the beta cell through a specific glucose transporter (GLUT-2), where it is phosphorylated by the key enzyme, glucokinase, to glucose-6-phosphate. Glucokinase is pivotal because it is the glucose-sensing mechanism of the beta cell. Further metabolism of glucose-6-phosphate eventually provokes increased intracellular ATP concentrations, which, in turn, induce changes in the membrane bound ATP-dependent K channel that allow for efflux of potassium, influx of calcium and, ultimately, exocytosis of insulin secretory granules. Thus, it is the metabolism of glucose within the beta cell that results in the subsequent secretion of insulin.

Figure 1. Intra Beta Cell Metabolism of Glucose.
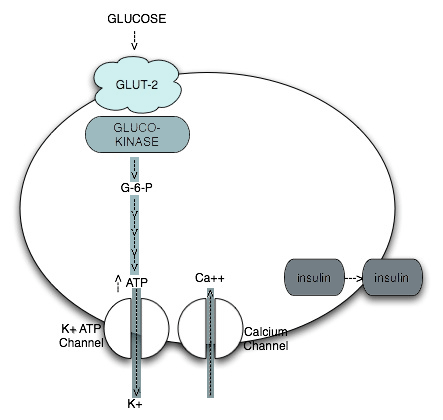
The concept of qualitative defects in beta cell function follows the recognition that the glucose signal to the beta cell, and the transmission of this signal through the various intracellular systems that ultimately result in insulin secretion, require a complex series of sequential steps, each of which is subject to dysfunction. For example, individuals with Type 2 diabetes may have a normal response to non-glucose secretagogues, such as arginine, implying that there are different signaling mechanisms for different secretagogues.
In regard to glucose-stimulated insulin secretion, it has long been known that not only patients with diabetes, but also those with impaired glucose tolerance, lose the ability to secrete insulin acutely in response to an intravenous glucose challenge (known as the 1st phase insulin response) while having an intact and, in fact, prolonged 2nd phase insulin response. Computer-driven glucose administration studies to create rhythmic oscillations in blood glucose levels have demonstrated a loss of synchronization between glycemia and insulin secretion in individuals with impaired glucose tolerance, which worsens in patients with diabetes.(2)
These are but a few examples of abnormalities in beta cell function that relate not to beta cell secretory reserve but rather to qualitative defects in beta cell action. Thus, it is likely that future directions in the treatment of Type 2 diabetes will exploit the expanding knowledge of the biology of the beta cell.
Incretins
The term "incretins" refers to secretory products of the intestine that influence beta cell function. The concept of incretins originated with the observation that the oral administration of glucose resulted in a greater insulin secretory response than the administration of intravenous glucose that produced a similar degree of glucose elevation. It was postulated that intestine-derived secretory products enhanced the ability of orally administered glucose to stimulate pancreatic beta cell secretion of insulin, a phenomenon termed the "incretin effect."
Subsequent studies demonstrated that the predominant intestine-derived secretory products that enhanced insulin secretion were glucose-dependent insulinotropic polypeptide (GIP) and glucagonlike peptide (GLP-1).(3),(4) Both GIP and GLP-1 are members of the glucagon peptide superfamily and share considerable structural homology. However, their effects on mechanisms related to glucose homeostasis differ considerably.
GIP
GIP is a 42-amino acid peptide, derived from a larger 153-amino acid precursor, ProGIP, that is secreted by the K-cells of the intestine located primarily in the duodenum and proximal jejunum. Plasma levels of GIP are low in the fasting state but rise within minutes postprandially.
Although the initial observation of GIP action related to the inhibition of gastric acid secretion (resulting in its original designation as a "Gastric Inhibitory Polypeptide"), this effect was seen predominantly at supraphysiologic dosages. GIP was later found to have effects on insulin secretion at physiologic concentrations; GIP administration amplified glucose-stimulated insulin secretion but did not significantly affect insulin secretion when glucose levels were not elevated.(3) This resulted in the change in terminology to Glucose-dependent Insulinotropic Polypeptide, still abbreviated as GIP. The glucose-dependent insulin secretory effect was seen only in the presence of hyperglycemia such as in the postprandial state. GIP was also found to have a variety of effects on fat metabolism in adipocytes, including lipoprotein lipase activity and fatty acid synthesis.(3)
Despite the demonstration that GIP plays a significant role in the regulation of insulin secretion in nondiabetic animals, GIP has not been effective therapeutically in animal models of diabetes. In these diabetic animals, GIP levels are not deficient and exogenous GIP administration is, compared to nondiabetic animals, actually less insulinotropic. Similarly, GIP levels are normal or slightly increased in humans with Type 2 diabetes. Studies of GIP infusion in humans have been inconsistent but, in general, have not shown a significant positive effect on insulin secretion in Type 2 diabetic patients. This may, once again, reflect the observation that GIP, although important in the euglycemic state, has little incremental effects on insulin secretion when glucose concentrations are above 140 mg/dl.
GLP-1 exists in two forms, a 30- and a 31-amino acid peptide, both of which are derived from the proglucagon precursor, a larger polypeptide that includes the sequence for glucagon as well as other peptides.
Figure 2. Proglucagon
| GRPP | GLUCAGON | IP-1 | GLP-1 | IP-2 | GLP-2 |
GRPP = glicentin-related pancreatic peptide
IP1 = Intervening Peptide 1
IP2 = Intervening Peptide 2
GLP1 = Glucagon-Like Peptide 1
GLP2 = Glucagon-Like Peptide 2
GLP-1 is secreted by the L-cells of the intestine, which are principally located in the ileum and colon. Despite the distal location of these cells, GLP-1 secretion occurs within minutes of food ingestion. This rapid response to food ingestion implies that a neural mechanism is responsible for the early secretion, whereas direct contact of the L-cells with nutrients may be responsible for later secretion. As with GIP, GLP-1 levels are low in the fasting state, but rise postprandially, and GLP-1 also promotes glucose-dependent insulin secretion. Unlike GIP, GLP-1 effects on insulin secretion are preserved in patients with Type 2 diabetes, thus offering the potential for therapeutic applications. Both peptides have a short circulatory half-life (5-7 minutes for GIP, and 2 minutes for GLP-1). The peptides are metabolized by the enzyme dipeptidyl peptidase-IV (DPP-IV).
In addition to its actions enhancing glucose-dependent insulin secretion, GLP-1 has also been found in animal studies to have other effects on pancreatic beta cell function and structure.(5) Effects include:
- Stimulation of insulin biosynthesis
- Differentiation of pancreatic precursor cells into beta cells
- Proliferation of beta cells
- Reduction in beta cell apoptosis (cell death).
The net result of these actions is to increase the mass of beta cells and, thus, increase insulin secretory capacity.
GLP-1 Actions
- Amplification of glucose-stimulated insulin secretion
- Stimulation of insulin synthesis
- Promotion of differentiation of beta cell precursors
- Inhibition of beta cell apoptosis
- Inhibition of glucagons secretion
- Reduction in rate of gastric emptying
- Inhibition of appetite
GLP-1 has other effects that may limit the degree of hyperglycemia. Patients with diabetes typically have elevated fasting glucagon levels, and they also do not suppress glucagon appropriately in the postprandial state. Glucagon increases blood glucose levels through the stimulation of hepatic glucose production via stimulation of glycogen breakdown as well as gluconeogenesis. GLP-1 inhibits glucagon secretion from the pancreatic alpha cells and, as such, serves a complementary function to insulin in the regulation of postprandial glycemic excursions.
As with its action on the beta cell, the inhibition of glucagon secretion by GLP-1 is glucose-dependent. The glucose dependency of GLP-1 action is critical to the avoidance of hypoglycemia. As glucose levels rise, insulin secretion is enhanced and glucagon secretion is suppressed. However, as glucose levels fall, these effects no longer occur, thus preventing drops in glucose levels below normal. In essence, the incretins do not directly stimulate insulin secretion but instead enhance the ability of glucose to stimulate insulin secretion.
GLP-1 Effects on Nutrient Intake and Digestion
An underappreciated phenomenon in patients with diabetes is the impact of hyperglycemia, per se, on rates of gastric emptying. Gastric motility studies in patients with diabetes have found that most have accelerated gastric emptying.(6) The accelerated emptying can result in both an increase in the rate of absorption of dietary carbohydrate and a loss of synchronization between nutrient availability and insulin secretion. Together, these factors produce an exaggerated postprandial hyperglycemia. GLP-1, however, can delay gastric emptying, which then tends to limit the rate of rise in glucose levels after meals. Additionally, GLP-1 directly inhibits appetite through the central nervous system.(7) These varied effects thus complement the ability of GLP-1 to enhance insulin secretion.
Patients with Type 2 diabetes have a defective incretin effect as evidenced by diminished insulin secretion in response to meals.(8) More recently, it has been demonstrated that patients with Type 2 diabetes also have a reduced GLP-1 response to meals.(9) Thus, the defect in nutrient-induced insulin secretion appears to be, at least in part, related to deficiency in GLP-1 secretion. As mentioned previously, patients with diabetes are also known to have an absent 1st phase insulin secretory response to intravenous glucose. Studies have indicated that administration of GLP-1 to patients with diabetes restores this 1st phase of insulin secretion.(8)
Therapeutic Implications
The recognition of the key role played by GLP-1 in glucose regulation has led to attempts to develop drug therapies that replace, mimic or enhance GLP-1 activity.(10) Because of its short plasma half-life (less than two minutes), native GLP-1 is effective only when given by continuous infusion. In a six-week study of 20 patients with Type 2 diabetes, Zander et al. administered GLP-1 by continuous subcutaneous infusion.(11) Over that relatively brief period of time, mean Hemoglobin A1c levels dropped by 1.3%, accompanied by a 77 mg/dl drop in fasting glucose. Consistent with the effects described earlier, gastric emptying was inhibited and a 1.9 kg weight loss was observed. Thus, this proof of concept study demonstrated the efficacy of GLP-1 replacement in patients with diabetes.
There have been two lines of drug development research aimed at capitalizing on the effectiveness of GLP-1 to improve glycemic control in diabetes: 1) the development of analogs to GLP-1 which can function as GLP-1 receptor agonists but are resistant to the activity of the degrading enzyme DPP-IV, and 2) the development of inhibitors of DPP-IV, thereby delaying the degradation of endogenous GLP-1.
Exenatide, a naturally occurring GLP-1 receptor agonist, was originally isolated from the saliva of the Gila monster. It has 53% homology with human GLP-1 but is resistant to degradation by DPP-IV. Exenatide's half-life is 26 minutes, compared to two minutes for the native compound. It is administered by subcutaneous injection, typically at a dose of 10 mcg BID.
Studies with exenatide as monotherapy, or in combination with either sulfonylureas, metformin or combination sulfonylurea/metformin, have all demonstrated consistent effects on glucose lowering. For example, Buse et al. demonstrated that among patients deemed sulfonylurea failures, with an average baseline Hemoglobin A1c of 8.6%, the addition of exenatide resulted in a drop of 0.86 in HbA1c accompanied by a 1.6 kg weight loss.(12) Those patients with the highest HbA1c levels at study entry had the greatest response to drug therapy (Figure 3).
Figure 3. Glucose Lowering With Exenatide
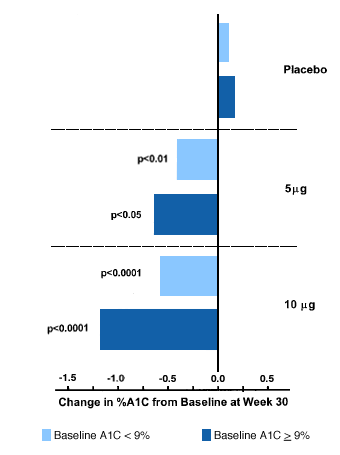
Adapted from Buse et al.
The remarkable weight loss in the face of improved glycemic control is in contrast to the weight gain typically seen with insulin, as well as with most oral antidiabetic medications. In a 28-day study of patients treated with exenatide who had been on metformin and/or sulfonylurea therapy, Fineman described HbA1c reductions of 0.7-1.1 with no weight gain, compared to a 0.9 kg weight gain seen in the placebo group.(13) These beneficial effects on weight are apparently related to the centrally mediated effects of GLP-1 agonists on food intake. Exenatide was approved for marketing in the United States in April 2005 and is the only drug in this category currently available.
Liraglutide, another injectable GLP-1 receptor agonist, is currently in Phase 2 clinical trials. Liraglutide, which has been chemically modified by fatty acid acylation to reduce the rate of enzymatic degradation, has 97% homology with native GLP-1. Its plasma half-life is 12 hours and short-term studies have demonstrated beneficial effects in Type 2 diabetes.(10) Both exenatide and liraglutide provoke early, and typically transient, nausea but are otherwise well tolerated.
Alternatively, endogenous GLP-1 activity can be enhanced through the use of inhibitors of DPP-IV activity. Several orally active DPP-IV inhibitors are currently in clinical trials. These agents reduce degradation of both GLP-1 and GIP, so the resulting glycemic benefits probably relate to enhanced activity of both incretins. Although the presence of deficiency in GLP-1 secretion in Type 2 diabetes might be expected to limit the overall efficacy of this class of drug, results of Phase 2 clinical trials have revealed clinically significant reductions in Hemoglobin A1c with chronic administration. The relative efficacy of this approach, compared to the use of GLP-1 receptor agonists, remains to be seen.
Summary
Drug therapies which restore the incretin effect represent a renewed focus on beta cell dysfunction as a major cause of the evolution of glucose intolerance in individuals with insulin resistance. Continued research on the impact of GLP-1 on the preservation and/or expansion of beta cell mass offer the potential for this category of medications to alter the natural history of Type 2 diabetes otherwise characterized by the progressive loss of beta cell function.