Course Authors
Antigoni Triantafyllopoulou, M.D., and Peter Barland, M.D.
Release Date: 03/21/2005
Upon completion of this Cyberounds®, you should be able to:
Describe the glandular and systemic manifestations of primary Sjogren's syndrome
Differentiate primary Sjogren's syndrome from other diseases that cause sialadenitis and keratoconjunctivitis (dry mouth and dry eyes)
Identify the prognostic factors that may produce increased mortality in patients with primary Sjogren's syndrome
Describe the clinical and laboratory criteria needed to make a diagnosis of primary or secondary Sjogren's syndrome
Discuss the current therapeutic modalities for the management of patients with Sjogren's syndrome.
Sjogren's syndrome (SS) is characterized primarily by dryness of the mouth (xerostomia) and eyes (xeropthalmia) resulting from a chronic lymphocytic inflammation of the salivary and lacrimal glands.(1)
Figure 1. Salivary Gland.
Patients with SS have a very high prevalence of circulating autoantibodies and the infiltrating lymphocytes appear to be immunologically activated. These findings, together with co-existent connective tissue disease, support an autoimmune etiology. However, a recent report of coxsackievirus genes and capsid protein detected in the salivary glands of patients with SS raised the possibility that a viral infection may also play a role in the pathogenesis of SS.(2)
Epidemiology
SS is one of the most common autoimmune diseases with a 1-3% prevalence in the general population.(3) This prevalence is equal to or higher than the rate for rheumatoid arthritis (RA), yet SS remains undiagnosed more than half of the time. Women are affected much more often than men (ratio of 9:1). The syndrome can affect patients at any age; however, it is mostly prevalent in women in their thirties or forties.
It is important to establish a diagnosis of SS for a number of reasons:
- The quality of life of patients with SS is significantly affected. Diagnosis and treatment may help alleviate their symptoms and prevent further morbidity.
- A subgroup of patients with SS is at increased risk for the development of lymphoma. This subgroup can be identified at the earliest stages of the disease, if a diagnosis of SS has been made.
- SS may be accompanied by severe systemic manifestations, such as systemic necrotizing vasculitis and membranoproliferative glomerunonephritis, that may require aggressive immunosuppressive treatment to prevent end-organ damage. These are rare manifestations but important to recognize.
- The vast majority of SS patients will require no more than symptomatic treatment of xerostomia and xerophthalmia and non-aggressive management of their systemic manifestations (e.g., arthralgias, Raynaud's). These patients can be effectively managed by the general internist, with only occasional problem-based referral to specialized clinics.
Primary and Secondary SS
SS is classified as primary (pSS) when it occurs alone and secondary (sSS) when it is accompanied by another systemic autoimmune disorder such as systemic lupus erythematosus (SLE), RA or scleroderma. In addition, SS may often co-exist with organ-specific autoimmune disorders, e.g., Hashimoto's thyroiditis, and in this setting SS is often considered pSS.
pSS and sSS may differ in their manifestations based on which systemic autoimmune disease accompanies sSS. For example,
- In sSS + RA, patients typically present primarily with eye dryness and lack the systemic manifestations (described below) that are characteristic of pSS. The minor salivary gland biopsy, though, is indistinguishable from pSS.
- In sSS + SLE, patients may have many systemic manifestations but the minor salivary gland biopsy is different from pSS: lymphocytic infiltrates are perivascular in sSS with SLE and periductal in pSS.
Manifestations of SS
As shown in Table 1, manifestations can be thought of as:
- Glandular, which result from destruction of exocrine glands from lymphocytic infiltrates and
- Systemic, which do not have a glandular pathology (thus are often referred to as extraglandular) and are characterized by lymphocytic infiltration of the skin, lungs, kidneys, peripheral nervous system and by hematologic malignancies.
Table 1. Manifestations of Sjogren's Syndrome.
Glandular Manifestations
- Xerostomia (dry mouth)
- Parotid gland enlargement
- Xerophthalmia (dry eyes)
- Xerotrachea (dry trachea)
- Dyspareunia
Extraglandular Systemic Manifestations
- Fatigue
- Raynaud's phenomenon, palpable purpura, recurrent urticaria, annular erythema
- Arthralgias/arthritis
- Pulmonary involvement
- Esophageal dysmotility, early primary biliary cirrhosis, subclinical pancreatitis
- Interstitial nephritis with renal tubular acidosis or more rarely, glomerulonephritis
- Peripheral neuropathy
- Lymphoma
Glandular Manifestations
Xerostomia (from the Greek words xeros=dry and stoma=mouth, to describe destruction of salivary glands resulting in dry mouth): It is one of the two subjective diagnostic criteria of SS, along with xerophthalmia (see Table 4, "Diagnostic Criteria of SS"). Patients with xerostomia may complain of dry mouth, frequent need to drink fluids, or may simply complain of an unpleasant taste, difficulty eating dry food, soreness of the mouth and throat, or difficulty using dentures.
On exam, the usual pooling of saliva in the floor of the mouth is absent; in advanced disease, the oral mucosa appears dry and glazed and the surface of the tongue becomes red and lobulated, with partial or complete depapillation.
Since the antimicrobial function of the saliva is lost (normally, salivary glands produce 1-1.5L of saliva every day), patients with SS are prone to develop dental carries and increased rate of oral bacterial infections with Streptococcus mutans and Lactobacillus, as well as increased rate of oral candidiasis. Furthermore, SS is the most common underlying cause of acute bacterial infection of the salivary glands (sialadenitis) -- usually from staphylococcus or pneumococcus. With acute bacterial sialadenitis, patients present with tender swelling of the salivary gland and, sometimes, regional tender lymphadenopathy, fever and malaise.
Xerostomia is not specific for SS and can be found in a number of other conditions as listed in Table 2.
Parotid gland enlargement: This is common, occurring in 60% of patients with pSS. It may appear episodically or as chronic persistent enlargement. Parotid gland enlargement may present unilaterally but often becomes bilateral. In the patient who presents with unilateral parotid gland enlargement, it is important to consider the possibility of lymphoma or acute bacterial infection before attributing the swelling to SS [see differential diagnosis (D/D) of parotid gland enlargement in Table 2].
Xerophthalmia (destruction of the lacrimal glands resulting in dry eyes): This is the second of the two subjective criteria for the diagnosis of SS. A broader term for this condition is "keratoconjunctivitis sicca," denoting the clinical findings that result from destruction of corneal and bulbar conjunctival epithelia as the result of diminished tear secretion.
Patients with xerophthalmia will complain of itching, burning and a sensation of gravel or sand in their eyes. They may also complain of redness of their eyes, photosensitivity, eye fatigue and an ocular discharge. These symptoms are exacerbated in dry climates and air-conditioned environments or with exposure to cigarette smoke. Untreated, xerophthalmia may be complicated by corneal ulceration, vascularization, opacification and, more rarely, corneal perforation.
On exam, there may be dilation of the bulbar conjunctival vessels, pericorneal injection and lacrimal gland enlargement.
Keratoconjunctivitis sicca is also not specific for SS.
Other glandular manifestations: Destruction of the exocrine glands that line the oropharynx and the trachea may result in hoarseness and a chronic dry cough. Atrophic gastritis may result in hypochlorhydria, while dryness of the vagina can cause dyspareunia (though fertility is not thought to be affected). More than half of SS patients also complain of dry skin.
Extraglandular Systemic Manifestations
Approximately half of patients with pSS display systemic manifestations that may affect almost any organ system, thus making the diagnosis of SS difficult, especially if the systemic manifestations are the presenting symptoms and the glandular symptoms are mild or subclinical. In those cases, the diagnosis is still possible if the separate manifestations are recognized as part of the syndrome and appropriate laboratory work-up is pursued.
As shown in Table 1, systemic manifestations of pSS include:
- Fatigue: extreme, debilitating fatigue affects about 50% of pSS patients.
- Skin manifestations: besides dry skin, Raynaud's phenomenon affects about 30% of pSS patients and may characteristically precede sicca manifestations by many years. Other skin findings include small and medium vessel vasculitis involving the skin-- sometimes with purpura, ulcerations, recurrent urticaria and annular erythema of the face and trunk.
- Musculoskeletal manifestations: myalgias and polyarthralgias are common. Patients may develop nonerosive polyarthritis. Severe myositis is unusual.
- Pulmonary manifestations: these are common but seldom clinically significant. Patients will present with dry cough and may have dyspnea on exertion, which is rarely progressive. High resolution computed tomography may reveal subclinical interstitial lung disease in 30% of pSS patients and pulmonary function tests may show small airway obstruction.
- Gastrointestinal manifestations: esophageal dysmotility is common, while severe malabsorbtion from mucosal atrophy and lymphocytic infiltrates of the small intestinal wall has been reported rarely in pSS. Abnormal pancreatic tests usually are secondary to subclinical pancreatitis. Abnormal liver tests raise the possibility of hepatitis C which may cause a Sjogren's like syndrome with dry eyes and dry mouth [In this situation, however, the rest of the systemic manifestations of pSS are lacking and the pSS characteristic autoantibodies (anti-Ro and anti-La) are negative.]. Thus hepatitis C infection is considered an exclusion criterion for the diagnosis of pSS. A low-grade form of primary biliary cirrhosis is also associated with pSS and these patients have positive antimitochondrial antibodies.
- Renal involvement: interstitial nephritis with renal tubular acidosis (usually distal) is common, affecting about 7-9% of pSS patients, while membranoproliferative glomerulonephritis is rare.
- Neurologic manifestations: pSS may cause a peripheral neuropathy that is primarily sensory and may even be the presenting manifestation of the syndrome. Rarely, a severe painful sensory neuronopathy, secondary to damage to sensory neurons in the dorsal ganglia, has been reported. In these patients, anti-Hu antibodies have been found in the serum, along with the more characteristic antibodies of pSS. A focal demyelinating syndrome resembling multiple sclerosis may be associated with pSS, though this association is controversial.
- Hematologic/oncologic manifestations: Lymphomas in patients with pSS are usually of B-cell lineage and are of low- or intermediate-grade. They are primarily localized in extranodal areas such as the salivary glands and the gastrointestinal tract (called MALTomas- mucosa associated lymphoid tissue lymphomas). It is now known that pSS patients have a 44% higher relative risk of lymphoma compared to the general population and that 5% of pSS patients will develop lymphoma.
Laboratory findings that may help characterize the full clinical picture of a pSS patient are presented in Table 3.
Table 3. Laboratory Findings of Primary Sjogren's Syndrome.
- Circulating autoantibodies: e.g., anti-Ro and anti-La autoantibodies (detected in 40-60% of pSS patients, with anti-La being more specific for pSS rather than sSS), antinuclear autoantibodies, rheumatoid factor
- Polyclonal hypergammaglobulinemia (in 80% of patients with pSS)
- Mixed oligoclonal cryoglobulinemia (in ~20% of patients with pSS)
- Low C3 and/or C4 complement levels (consistently adverse prognostic factor)
- Electrolyte abnormalities and urinalysis consistent with renal tubular acidosis (hyperchloremic, hypokalemic acidosis with hyposthenuria), nephrocalcinosis (hypercalciuria) or rarely, glomerulonephritis (proteinuria, hematuria) in patients with SS and renal involvement
- Mildly elevated lipase, amylase with subclinical pancreatitis and pSS (rare)
- Positive antimitochondrial antibodies (7% of pSS patients) and, less frequently, abnormal liver function tests in patients with pSS and primary biliary cirrhosis
Clinical Predictors of Lymphoma (also clinical predictors of increased mortality) in pSS Patients
These predictors can be reliably identified within the first year of diagnosis of pSS. Depending on their presence or absence; it is now thought that pSS can be categorized as type 1 and type 2.(4),(5) Type 1 is the high-risk disease group of pSS that may develop lymphoma and type 2 comprises the rest (and majority) of pSS patients whose mortality is not affected.
The two clinical predictors of increased mortality are:
- Low C4 (and possibly low C3) complement levels;
- The presentation of palpable purpura.
These two predictors, along with parotid gland enlargement, were able to predict almost all future lymphoproliferative disease diagnoses in a prospective cohort study of 723 pSS patients.(4)
Revised Criteria for the Diagnosis of SS
In 2002, an American-European consensus group proposed revised diagnostic criteria (see Table 4).
Table 4. Diagnostic Criteria for Sjogren's Syndrome.
Two Subjective Criteria:
- Ocular symptoms: persistent dry eyes (> 3 months)
- Oral symptoms: persistent dry mouth
Four Objective Criteria:
- Signs of dry eyes: positive Shirmer's test or positive Rose Bengal score upon ophthalmologic evaluation
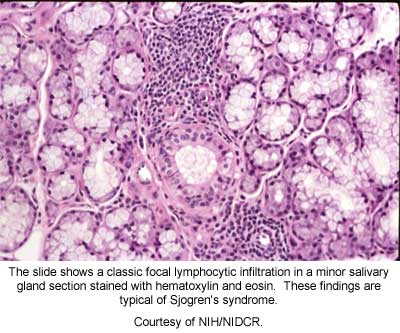
- Histopathology of lip biopsy: minor salivary glands with lymphocytic infiltrates that have a focus score more than 1, as measured by an experienced histopathologist
- Presence of either Ro or La autoantibodies in the serum
- Objective measurements of decreased salivary gland function (abnormal flow rate, scintigram or sialogram)
Exclusion Criteria:
- Hepatitis C infection
- AIDS
- Sarcoidosis
- Pre-existing lymphoma
- Past history of head and neck irradiation
- Graft vs. host disease
- Use of anti-cholinergic drugs
The presence of any 4 out of 6 criteria is indicative of the diagnosis of pSS with a sensitivity of 97% and a specificity of 89%. If 3 out of the 4 objective criteria are present, then pSS can be diagnosed with a sensitivity of 84% and a specificity of 95%, even if the patient doesn't complain of dryness. Presence of all 6 criteria had a sensitivity of 50% and a specificity of 100%.
sSS is diagnosed when 1 of the 2 subjective and 2 of the 4 objective criteria are present in a patient with another well-defined connective tissue disease.
These criteria and the studies that validated them stress the importance of objective tests such as Ro and La autoantibodies and histopathology for a sensitive and specific diagnosis of pSS.
Histopathology refers to the pathologic examination of minor salivary glands that are removed with a small (~1cm) incision of the mucosa overlying the lower lip. The size and number of lymphocytic foci that are adjacent to salivary ducts are counted and a focus score is measured. It is important to remember that other diseases such as sarcoidosis and chronic HCV and HIV sialadenitis may have a similar histopathology -- thus mandating an experienced pathologist to interpret the biopsy and keep in mind the full clinical picture of the patient.
Treatment
Treatment of glandular manifestations is symptomatic. The goal is to attempt to reduce future complications and improve quality of life:
- Xerostomia is treated with muscarinic agonists. Two are now available in the U.S.: pilocarpine hydrochloride, which is dosed as 5 mg tablets 4 times daily, and cevimeline hydrochloride, given as 30 or 60 mg tablets three times daily. Side effects may include excessive sweating (less often with cevimeline), nausea, rhinitis and diarrhea. Frequent dental visits are advised to prevent and treat dental carries and oral infections. Patients with SS should avoid, if possible, tricyclic antidepressants, beta blockers and diuretics, since these may worsen their symptoms.
- Xerophthalmia is treated primarily with topical hydrophilic solutions to prevent complications such as corneal ulceration. Temporary occlusion of the puncta, through the insertion of plugs, or permanent occlusion, through electrocautery, are also employed to preserve existing tears. Pilocarpine and cevimeline may also be used for xerophthalmia. Topical cyclosporine A has recently been reported as effective for xerophthalmia.
Treatment of systemic manifestations is usually centered on non-aggressive symptomatic treatment with NSAIDs or even low-dose corticosteroids or hydroxychloroquine for arthralgias. Long-term immunosuppression has not been shown to be of benefit and is hazardous, especially since the syndrome usually does not carry an increased mortality. Moderate doses of corticosteroids may be used in ulcerated severe vasculitic skin lesions or with renal tubular acidosis that is resistant to replacement therapy and in the initial management of membranoproliferative glomerulonephritis.
Conclusions
SS is a common chronic autoimmune disorder that may present with numerous manifestations. A diagnosis of the syndrome can be made if there is subjective evidence of eye or mouth dryness along with objective findings of reduced tear and/or saliva secretion, and, most importantly, positive Ro or La autoantibodies or positive minor salivary gland histopathology (from lip biopsy). The presence of low complement levels and palpable purpura within the first year of diagnosis of pSS are predictive factors of increased mortality and B-cell lymphoma. In the majority of patients, those factors are absent, their disease course is benign and treatment is mostly symptomatic.