Course Authors
Bruce S. McEwen, Ph.D.
Release Date: 06/15/2004
Upon completion of this Cyberounds®, you should be able to:
Describe three brain areas involved in processing stressful events, and describe how acute stress affects their function
Describe what chronic stress does to these brain areas and how excitatory amino acids are involved along with circulating stress hormones
Describe how chronic stress affects behavior and memory in animal models.
We use the word "stress" to refer to experiences that threaten us and push us beyond our ability to cope physically and psychologically with a challenge that confronts us. Stressful experiences activate the secretion of hormones, such as cortisol and adrenalin, and turn on neurotransmitter systems in the brain, including the ubiquitous excitatory amino acid transmitters (EAA). The EAA, together with cortisol and adrenalin, mediate adaptive processes that improve our ability to cope with the challenging situation both physically and psychologically, and NMDA receptors play a key role.
Repeated stress causes measurable structural changes in key brain areas, such as the amygdala, hippocampus and prefrontal cortex that alter the ability of these brain regions to function in processing fear and anxiety and learning and memory. These brain areas also influence the output of hormones and other mediators that affect the functioning of cardiovascular, metabolic and immune systems. Repeated stress produces brain and body pathophysiology similar to the effects seen in people with mood and anxiety related disorders such as depressive illness.
Key roles of the Hippocampus, Amygdala and Prefrontal Cortex
The hippocampus, amygdala and prefrontal cortex contain receptors for adrenal steroids, which work in concert with EAA and other neurotransmitter systems, that regulate excitability and structural changes. The amygdala mediates anxiety, fear and fear-related learning and has important outputs to the autonomic nervous sytems and neuroendocrine system. The amygdala works with the prefrontal cortex and hippocampus to encode and recall experiences related to "context," for example, where you were and what you were doing when a traumatic event happened, such as the terrible events of Sept. 11, 2001. The prefrontal cortex provides the basis for short-term "working" memory and also plays a key role in extinction of learned fear, which is a form of new learning overlaid on top of the original fear conditioning. The hippocampus provides the memory of "context" as well as declarative, episodic and spatial memory.
Role of Stress Mediators in Behavioral Adaptation
Acutely, after a stressful event, the amygdala and hippocampus are both involved in contextual fear conditioning and in passive avoidance learning, i.e., learning to be inactive in order to avoid a painful experience. In fear conditioning, glucocorticoids (e.g., cortisol) enhance learned fear(18) and they play an important role in forming the memory of where you were and what you were doing in contextual fear conditioning, but glucocorticoids do not influence the actual effect of footshock in rats that are already familiar with the context where the shock is administered.(69),(70) This suggests that the hippocampal role in contextual fear conditioning is enhanced by moderate levels of glucocorticoids, but that the fear conditioning is either not so dependent on glucocorticoids or is so strong that glucocorticoid influences are hard to demonstrate.
Yet there is evidence for an influence of glucocorticoids on the flow of information within the amygdala. Glucocorticoids potentiate serotonin-inhibition of the processing of excitatory input to the lateral amygdala from the thalamus, suggesting that there is a mechanism for containing, or limiting, the sensory input that is important for fear conditioning.(97) Thus adrenal steroids may regulate the nature of the signals that reach the amygdala and allow for greater discrimination of the most salient cues for learning.
Moreover, in passive avoidance, both catecholamines and glucocorticoids play a role in facilitating the learning.(7),(77) Catecholamines work outside of the blood-brain barrier and their effects can be blocked by beta adrenergic blocking agents that do not cross the blood brain barrier.(7) Glucocorticoids enter the brain, and local implants of exogenous corticosterone into hippocampus, amygdala and nucleus tractus solitarii (NTS) are all able to enhance passive avoidance learning.(77)
Adrenal steroids also play a supporting role in the learning of a spatial navigation task in mice.(63) Adrenalectomy impairs the acquisition of the memory of hidden platform location in the Morris water maze, and glucocorticoid administration restores the normal learning curve; however, in mice in which the glucocorticoid receptor was deleted and replaced with a glucocorticoid receptor (GR) that lacks the DNA binding domain, glucocorticoids have no effect to improve task acquisition.(63) This finding illustrates a role for glucocorticoid receptors acting upon the genome in a task that is known to depend on the hippocampus. Interestingly, other actions of glucocorticoids via glucocorticoid receptors are known to involve the protein-protein interactions that are not prevented in mice carrying the GR defective in the DNA binding domain.(76)
Other evidence for glucocorticoid actions supports an inverted U-shaped dose-response curve in which low to moderate levels of adrenal steroids enhance acquisition of tasks that involve the hippocampus, whereas high levels of glucocorticoids disrupt task acquisition.(15),(21),(22),(70) Adrenal steroids have biphasic effects upon excitability of hippocampal neurons that may underlie their biphasic actions on memory and recall.(21),(54),(64),(65),(65),(66) The exact role of adrenal steroids in the prefrontal cortex is unknown at this time, although it probably is similar to its role in hippocampus and amygdala.
Adaptive Structural Plasticity In Key Brain Areas
When stress becomes chronic, the brain adapts and changes structurally. These changes alter behaviors involving memory, fear and aggression. Repeated stress causes neurons to be remodeled in the prefrontal cortex and hippocampus with dendrites becoming shorter and less branched; as a result, learning and memory that depend on these brain structures are impaired.(54),(71) In the amygdala, dendrites grow and new synapses are formed in key regions that are involved in the processing of fear and fear learning. The consequences of these changes include increased fear and aggression.(16),(104) In the dentate gyrus of the hippocampus, where new neurons are born during adult life, chronic stress reduces neurogenesis and shrinks the size of the dentate gyrus(54),(89),(104),(109)
As far as we know, most of these changes are reversible as long as the stressor is terminated after weeks; however, it is possible that there is a long-lasting imprint on the brain that makes it more vulnerable to later stressful events. This will be considered in a later Cyberounds® in this series. Meanwhile, it is important to understand the mechanism by which structural plasticity is produced by chronic stress, and, therefore, we shall spend some time on the hippocampal formation because it represents the best-studied area.
Unique anatomy of the hippocampus makes it vulnerable: Within the hippocampus, the input from the entorhinal cortex to the dentate gyrus is ramified by the connections between the dentate gyrus and the CA3 pyramidal neurons. One granule neuron innervates, on the average, 12 CA3 neurons, and each CA3 neuron innervates, on the average, 50 other CA3 neurons via axon collaterals, as well as 25 inhibitory cells via other axon collaterals.(54) The excitatory neurotransmitter in these connections is glutamate and the inhibitory neurotransmitter is GABA.
Figure 1. Why is the CA3 So Vulnerable?
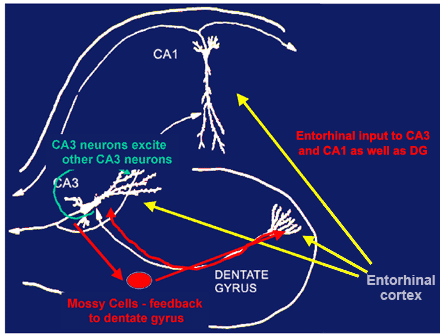
Feed-forward excitability serves memory functions but increases vulnerability for excitotoxicity.
Feed forward and feedback innervation of CA3 from the dentate gyrus is responsible for the unique role of the DG-CA3 system in memory of sequences of events. However, it also makes the CA3 region particularly vulnerable to excitotoxic damage and the effects of chronic stress.
The net result is a 600-fold amplification of excitation, as well as a 300-fold amplification of inhibition, that provides some degree of control of the system. As to why this system exists, the dentate gyrus--CA3 system is believed to play a role in the memory of sequences of events, although long-term storage of memory occurs in other brain regions.(25),(43)
Neurogenesis in the dentate gyrus: There is structural plasticity within the DG-CA3 system, as new neurons continue to be produced in the dentate gyrus throughout adult life(32) and CA3 pyramidal cells undergo remodeling of their dendrites,(54) as will be discussed further below.(56) The sub-granular layer of the dentate gyrus contains cells that have properties of astrocytes (e.g., expression of glial fibrillary acidic protein) and which give rise to granule neurons.(82) After administration of a precursor (BrdU) to label DNA of dividing cells, newly-born cells appear as clusters in the inner part of the granule cell layer, where a substantial number of them will go on to differentiate into granule neurons within as little as seven days. The new granule neurons appear to be quite excitable and capable of participating in long-term potentiation. In the adult rat, 9000 new neurons are born per day and survive with a half-life of 28 days.(2)
There are many hormonal and neurochemical modulators of neurogenesis and cell survival in the dentate gyrus.(1),(19),(32),(51),(101) Neurogenesis in the adult dentate gyrus is enhanced by the circulating hormone, IGF-1, and by the neurotransmitter, serotonin, and by a number of antidepressant drugs. IGF-1 is the mediator of the ability of exercise to increase cell proliferation in the dentate gyrus. Lack of IGF-1 and insulin in diabetes has the opposite effect and decreases cell proliferation.
Neurogenesis and/or survival of newly-born cells are increased by putting mice in a complex ("enrichred") environment.(37) It is increased by a form of classical conditioning that activates the hippocampus ("trace conditioning") and prolongs the survival of newly-born dentate gyrus (DG) neurons.(30),(88) On the other hand, certain types of acute stress and many chronic stressors suppress neurogenesis or cell survival in the dentate gyrus, and the mediators of these inhibitor effects include excitatory amino acids acting via NMDA receptors and endogenous opioids.(9),(26),(31),(54)
NMDA receptors play a particularly important role - for example, NMDA receptor antagonist treatment increases neurogenesis in the dentate gyrus in young and old rats, alike, and leads to 20% increases in neuron number.(8),(62) Chronic stress has equally potent effects on neurogenesis and neuronal survival, operating through elevated excitatory neurotransmission along with elevated glucocorticoids. Chronic restraint stress (CRS) for 21d suppressed neurogenesis and CRS for 42d causes the number of DG neurons to decrease along with total DG volume.(68)
Figure 2. Cell Proliferation in the Dentate Gyrus.
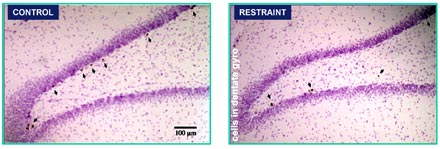
A single restraint stress does not suppress cell proliferation.
Repeated restraint stress for 21d suppresses cell proliferation.
Repeated restraint stress for 42 days reduces volume of DG and DG neuron number.
Cell proliferation in the dentate gyrus, reflecting incorporation of BrdU into DNA, is shown for control and 21d restraint stress animals. From Pham et al 2003.
Remodeling of dendrites: Another form of structural plasticity is the remodeling of dendrites in the hippocampus.(56) CRS causes retraction and simplification of dendrites in the CA3 region of the hippocampus.(56)
Figure 3. CA3 Pyramidal Cells Show Dendritic Shrinkage.
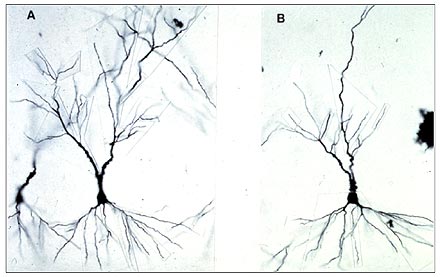
Shorter dendrites have been found in DG and in CA1 as well as CA3 after both repeated stress and chronic corticosterone treatment.
Such dendritic reorganization can also be seen in rats undergoing adaptation of psychosocial stress in the visible burrow system (VBS). The VBS is an apparatus with an open chamber where there is a food and water supply, as well as several tunnels and chambers.(4) Rats can be observed from above by a video camera in this apparatus. In the VBS, male rats housed with several females establish a dominance hierarchy within several days. Over the course of the next week, a few subordinate males may die and others (with scars from bite marks) will show enlarged adrenals, low testosterone and many changes in brain chemistry. The dominant rat shows the fewest scars and has the highest level of testosterone but also has somewhat larger adrenal glands than cage control rats.
Regarding changes in brain structure, it was the dominant rat that had a more extensive pattern of debranching of the apical dendrites of the CA3 pyramidal neurons in the hippocampus, compared to the subordinate rats, which showed reduced branching compared to the cage controls.(57) What this result emphasizes is that it is not adrenal size or presumed amount of physiological stress per se that determines dendritic remodeling, but a complex set of other factors that modulate neuronal structure. We refer to the phenomenon as "dendritic remodeling" and we generally find that it is a reversible process. In hibernating hamsters, it occurs in a matter of hours and reverses itself just as quickly when hibernating animals are aroused from torpor (Magarinos, McEwen, Pevet, unpublished). Below we consider mechanisms involved in structural remodeling.
The role of adrenal steroids in the structural remodeling described above reflects many interactions with neurochemical systems in the hippocampus, including serotonin, GABA and especially excitatory amino acids.(54)
Figure 4. Dendritic Remodeling: Where Do Glucocorticoids Work?
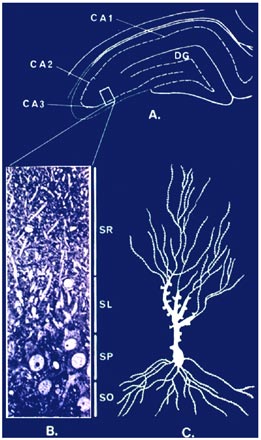 |
|
Probably the most important interactions are those with excitatory amino acids such as glutamate. Excitatory amino acids released by the mossy fiber pathway play a key role in the remodeling of the CA3 region of the hippocampus, and regulation of glutamate release by adrenal steroids may play an important role.(36),(44),(45),(107) We have found that the glutamate transporter, Glt-1, is elevated by CRS in hippocampus, particularly in the CA3 region, providing another indication that elevated glutamate levels are an important component of structural plasticity.(75) We previously showed that NMDA receptor blockade and the Na/Ca channel blocker, phenytoin, both block, CRS- and glucocorticoid-induced remodeling of dendrites in CA3.(48),(49),(106) Recent evidence indicates that presynaptic receptors containing kainate receptor subunits, such as GluR6, are important for the feed-forward actions of glutamate on mossy fiber terminals(17) and one study showed that a number of kainate receptor subunit mRNA's are regulated biphasically by adrenal steroids.(36) In particular, preferential mineralocorticoid receptor occupancy by low corticosterone (CORT) levels enhanced mRNA levels for KAR2, GluR6 and GluR7.(36) This agrees with the finding that MR activation by aldosterone in ADX rats restored levels of 3H kainate binding in the mossy fiber region of CA3.(107) However, further studies are needed.
Implications for Mood and Anxiety-Related Psychiatric Disorders
Stress is widely acknowledged as a predisposing and precipitating factor in psychiatric illness.(11)(38) Stress hormones are elevated in major depressive illness, and this is due to distortions in the diurnal rhythm.(79) Normally low evening levels of cortisol are increased in depression (20),(110) and the stress hormone axis in major depression is frequently found to be resistant to suppression by the synthetic glucocorticoid dexamethasone.(10) In major depression and a number of other mood and anxiety disorders, there are reports of hippocampal and prefrontal cortex volume loss and enlargement of the amygdala.(55),(83) Structural remodeling in these brain regions is important for human psychiatric disorders because the altered circuitry is likely to contribute to impaired cognitive function and affect regulation.
The hippocampal volume loss in major depressive illness is related to duration of the depression rather than to age per se of the patients.(6),(86),(87) Not all studies report such changes,(102),(78) and the reasons for these different results are beyond the scope of this discussion, but they may be explained by differences in the duration of depression, as well as by gender and age. It should be noted that hippocampal size in elderly twins shows only 40% genetic contribution, with the predominant influence being environmental.(98) This emphasizes the potential importance of experiential factors and chronic wear and tear produced by glucocorticoids and excitatory amino acids in determining hippocampal volume.
In relation to geriatric depression, hippocampal atrophy has been noted in relation to depression in the elderly,(95) with an association detected with presence of the ApoE4 genotype.(39) In subjects with a long-term history of depression, Sheline and colleagues described, in their MRI images, evidence for discontinuities that might represent sites of damage.(87) Although some recent postmortem studies on brains from depressed individuals did not show neuron loss in the hippocampus,(46),(59) the duration of the depression and the subtype of depression were not carefully controlled. Thus the possibility that neural damage may ultimately occur in major depression cannot be disregarded, particularly when depression lasts a long time. However, in a recent study in young depressed subjects, hippocampal volume was not smaller in first episode depression but declined rapidly over several years.(47) The key unanswered question is whether such changes can be prevented or even reversed. In Cushing's disease, where there is cognitive impairment and hippocampal volume loss, correction of the hypercortisolemia resulted in at least partial reversal of hippocampal shrinkage over several years.(5),(91),(92)
It is important to note that other brain regions besides the hippocampus are affected in depressive illness and undergo structural changes. One region is the prefrontal cortex, and structural imaging(23) showed loss of volume in familial pure depressive disorder, whereas autopsy studies (72),(73),(74) have shown loss of volume and glial cells, as well as neuronal density in both unipolar and bipolar disorder. Recent animal model studies demonstrate that chronic glucocorticoid treatment and CRS induce remodeling of dendrites in the rat medial prefrontal cortex.(109),(71) However, much more work needs to be done on this brain region.
Depressive illness is associated with a hyperactivation of the amygdala(24),(84) and, more recently, with an actual enlargement of the amygdala in the first episode of major depression.(28) This is reminiscent of the increased dendritic branching reported in rats after repeated immoblization stress [see above and (12)]. Since the amygdala integrates information related to fear and strong emotions, and also sends outputs via the central nucleus for autonomic arousal and via the basal nucleus for more active aspects of coping,(42) the elevation of amygdala activity may be a first step that leads to overactivation of systems involved in physiological and behavioral coping.
Structural remodeling of the brain in chronic stress in animal models offers insight into brain changes that take place in anxiety and mood disorders, which are frequently precipitated by stressful life events. Studies of one of the animal models of chronic stress, the tree shrew, revealed that treatment with antidepressant, anti-seizure and mood-stabilizing drugs prevents stress induced hippocampal structural changes, as well as relieves symptoms in the animals that are reminiscent of depression.(19),(50),(103) Besides reduced neurogenesis in DG, there is also evidence for reduced size of principal neuron cell bodies in the hippocampus, which is consistent with reduced size of the dendritic tree.(96) Taken together, such structural changes seem likely to play a major role in the volume loss in the human hippocampus and the related effects on cognitive function and affect.(83)
How the Brain and Body Interact: Systemic Consequences in Depression
When mood and anxiety disorders last for months to years, there is wear and tear on the body that results in a number of pathophysiological consequences. One reason for this is that the amygdala, which becomes hyperactive in depression, regulates both autonomic nervous system activity and ACTH and cortisol production through outputs of its central nucleus.(42),(80) It is important to note that there are reports that in recurrent major depression of long duration, the amygdala may undergo shrinkage.(85),(86) It is thus possible that initial hypertrophy gives way to atrophy in this important brain structure.
Besides the brain changes in major depression, there are other changes in the body that reflect dysregulated hypothalamus-pituitary-adrenal axis (HPA) and autonomic activity and are slow in developing. These produce cumulative pathophysiology, which may also be reversible if caught in time. Such cumulative, long-term effects include bone mineral loss(13),(58),(81) and abdominal fat deposition.(52),(99),(108) Moreover, the combination of long term pathophysiology, together with dysregulation of the autonomic nervous system activity in major depression,(100) is associated with increased blood platelet reactivity(40),(61),(105) and increased risk for cardiovascular disease.(3),(35),(60),(67)
There are parallels between the story for major depression and what is known about psychiatric and somatic features of Cushing's disease involving melancholia, depression, abdominal obesity, bone mineral loss and increased risk for cardiovascular disease.(14),(29),(93),(94) We have also noted that there is evidence for hippocampal atrophy in Cushing's Disease along with memory impairments.(27),(53),(90)
Finally, a largely unexplored area concerns the effects of antidepressant medication on the brain and body changes associated with depressive illness. On the one hand, certain antidepressants may contribute to some of the associated pathophysiology, such as cardiovascular instability.(41) On the other hand, withdrawal from antidepressant treatment may cause imbalances in neurotransmitter systems, with elevations of excitatory amino acid tone,(33), and contribute to the allostatic (i.e., chronic wear and tear) load that occurs as the depressive state continues.(33),(34)
Conclusion
The brain is capable of considerable adaptation and plasticity, and we have seen in another Cyberounds® of this series how excitatory amino acids and their receptors play an essential role in processes underlying learning and memory Stressful experiences promote learning of information needed to deal with a dangerous or threatening situation in the future and we have seen that excitatory amino acids, along with other neurotransmitters and circulating hormones, play vital roles in this adaptive plasticity. Chronic stress also causes adaptive plasticity that alters neuronal structure in brain areas, such as the amygdala, hippocampus and prefrontal cortex, changing behavior and the capacity to learn and remember.
Although many of these changes appear to be reversible, it is unknown to what extent they predispose the brain to potential damage to future stressors of the same or a different kind. What is clear is that the brain changes in response to repeated stress, changes aggressive and fear-related behaviors and the capacity to learn and remember. These alterations may promote greater vigilance and better preparedness for future danger. Yet, these same mechanisms appear to be co-opted by the brain when it is afflicted with depressive illness and other mood- and anxiety related disorders, and, in this case, the adaptive plasticity is likely to contribute to the pathophysiology of the disorder not only in the brain, but also in terms of the systemic consequences of the physiological dysregulation that results from prolongation of the psychiatric disorder.
The good news is that we are dealing with adaptive plasticity rather than outright neural damage. The hope is that the adaptive plasticity in these disorders may be treatable when we identify the right pharmacological tools and use adjunctive behavioral and psychotherapies. As we have seen, excitatory amino acids and their receptors are central to the brain mechanisms that are involved in this plasticity.