Course Authors
Simone Graber, Ph.D., Mary E. Morrison, Ph.D., and Shelley Halpain, Ph.D.
Release Date: 05/27/2004
Upon completion of this Cyberounds®, you should be able to:
Define dendritic spines and understand the concept of biochemical compartmentalization
Discuss the correlation between changes in spine shape/function and disease states
Discuss spine loss in the context of excitotoxicity.
Dendritic spines are micron-sized protrusions that form the postsynaptic part of synapses in the central nervous system that release the excitatory neurotransmitter glutamate. Spine-bearing neurons are found in the neocortex, striatum, hippocampus, cerebellum and other brain areas. Spines contain a specialized collection of molecules that enable the postsynaptic neuron to respond biochemically to glutamate, including different types of glutamate receptors,(1) structural proteins, and enzymes that transduce glutamate signals acting through intracellular calcium and other second messengers.(2),(3),(4)
Dendritic spines play critical roles in cognitive and motor function, as well as memory formation. Spines are lost or malformed in many disease states, including epilepsy, stroke, schizophrenia, mental retardation, dementia, and chronic drug and alcohol abuse. The purpose of this Cyberounds® is to review the structure, function, and development of spines, as well as to highlight molecular mechanisms for their regulation. In the future, such information may form the basis of new therapies for treating neurological and psychiatric diseases.
The Inner Universe
The human brain is estimated to contain more than 1013 dendritic spines.(5) These tiny structures were first noticed on cerebellar Purkinje neurons in Golgi-stained human brain preparations by the great neuroanatomist Santiago Ramon y Cajal in 1888,(6) and subsequently observed in many other brain regions. The small size of spines approaches the limits of optical resolution in light microscopy, so much of the early work that characterized spines in detail was conducted using electron microscopy. Most spines in the mature brain have a club-like appearance (Figure 1).
Figure 1. Computerized Reconstruction of a Dendrite Segment.
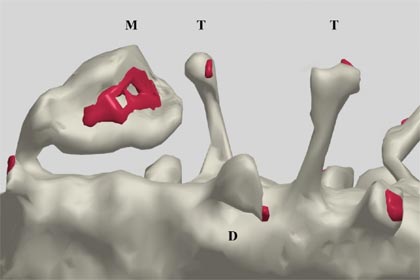
Computerized reconstruction of a dendrite segment ("D") visualized using serial section electron microscopy. Note the large variation in the shape and size of protrusions, including thin spines ("T"), and larger, mushroom-shaped spines ("M"). Red areas indicate the region occupied by the post-synaptic density (PSD), an organelle containing dense clusters of glutamate receptors, cell adhesion molecules, and signal transduction complexes (see text). Pre-synaptic terminals sit across from such PSDs, but are omitted from this figure to facilitate visualization of the spine morphology.
(Reprinted from Brain Research Reviews, Vol. 39(1), pp. 29, Fiala JC, Spacek J, Harris KM "Dendritic spine pathology: cause or consequence of neurological disorders?" copyright (2002), with permission from Elsevier.)
Spines are connected to their parent dendrite by thin stalks 0.04-1 μm long, and have variably shaped bulbous tips 0.5-2 μm in diameter.(7) Their density on mature, spiny neurons ranges from 1-10 spines per μm length of dendrite. The vast majority of CNS excitatory synapses contact spines, although such glutamate synapses can also form directly on the dendrite shaft (so-called "shaft synapses"). In contrast, nearly all inhibitory synapses are shaft synapses. Spiny neurons themselves can be either excitatory (such as pyramidal neurons in the neocortex and hippocampus) or inhibitory [such as the medium size projection neurons of the striatum or cerebellar Purkinje neurons, both of which release GABA (gamma aminobutyric acid)].
Spines Are Highly Dynamic
Early electron microscopic images of fixed brain preparations identified distinct morphological categories of spine shapes, such as "mushroom shaped," "thin," or "stubby."(8),(9) More recent experiments using living preparations and vastly improved light microscopy methods for fluorescence time-lapse imaging reveal that spine shapes are highly dynamic, and that a given spine shape may convert or "morph" into another on a time scale of seconds to minutes.(10),(11),(12) The function of this intrinsic motility is presently not known. Speculations include regulation of synapse adhesion, vesicle trafficking to control the concentration of glutamate receptors in the membrane, or changing the biochemical compartmentalization of the spine and thereby its signal processing parameters.(13)
Indeed, the wholesale appearance and disappearance of dendritic spines appear to be normal parts of brain function, not only for refinement of synaptic circuits during development, but also for synaptic remodeling in the adult. Such changes may be one way to encode memory.(14) Remarkably, new state-of-the-art imaging experiments in living mice have revealed that spines can turn over with half-lives of a day or less, even in adult animals.(15),(16) In somatosensory cortex, approximately 50% of spines were observed to turn over every few days, even though the overall numbers of spines remained constant.(15) However, in other parts of the adult cortex, spines appeared to be stable over many months, with only a small fraction being replaced in this time frame.(16)
As expected, both spine turnover and the rapid spine 'morphing' described above are greater in developing animals than in adults. Research is currently at an early stage in our ability to monitor spine turnover in vivo. It is nonetheless reasonable to postulate that the relative permanence of dendritic spines will be found to vary greatly depending on age, brain region, level of electrical and chemical activity, and disease state.
It is clear that the numbers of spines per dendrite segment can change under specific physiological circumstances. In vivo, significant cyclic changes in spine density are observed over the four-day estrous cycle in female rats(17),(18) and during hibernation and reawakening in ground squirrels.(19) Increasing the ratio of testosterone to estrogen by ovariectomy can increase synaptic density.(20) In addition, recent studies show that spines can appear very rapidly (within minutes) in response to physiological stimuli(21),(22) and collapse very rapidly (within minutes) in response to pathological stimuli.(23) These dynamic changes in spine shape and existence are regulated mainly through the actin, cytoskeleton, as discussed below.
Development of Spines
Studies of dendritic spines in developing humans, nonhuman primates, and rodents reveal a common overall pattern of spine development. Most of our current understanding of mechanisms comes from studies conducted using neuronal cultures from rodent brain tissue. Spine formation occurs as a late step in neuronal morphogenesis (e.g., postnatal week 2-3 in the mouse hippocampus). This timing and order of events is preserved in cultured neurons. Synaptogenesis begins as afferent axons reach their postsynaptic targets. At this initial stage, dendrites of nearly all neurons -- spiny and non-spiny -- bear numerous thin protrusions termed 'filopodia'.(24),(25) Dendritic filopodia are long, narrow protrusions that lack a bulbous head and contain lower amounts of F-actin than spines. They are more transient in nature than dendritic spines, extending and retracting from the dendrite shaft with half-lives of about 10 minutes, as compared to half-lives of hours, days or months for mature spines.(8),(25)
Gradually, over the course of several days, the numerous dendritic filopodia are replaced by spines. Spiny synapses have been proposed to arise by two mechanisms:
- synapse forms directly on the dendrite shaft, and a spine gradually grows out from the shaft
- a synapse forms on a dendritic filopodium and the filopodium gradually 'converts' into a mature spine.
Experimental evidence supports each of these mechanisms, and it seems likely that both can and do occur even within a single neuron.
Although there is direct experimental evidence for the involvement of filopodia in the formation of at least some dendritic spines, it seems clear that not all filopodia are destined to become spines.(25) Indeed, filopodia are also observed on non-spiny neurons early in their synaptogenesis phase.(24) It is likely that filopodia, with their high motility and transient nature, play key roles in establishing many kinds of synapses, because they maximize the chance encounter of pre- and post-synaptic elements during the search for synaptic partners. The emergence of a spine as a semi-permanent protrusion is probably not dictated by the presence or absence of a filopodium, but rather by the presence of molecular complexes that cluster and accumulate within the plasma membrane in response to signals from the nerve terminal.
Our current knowledge of molecular mechanisms in synaptogenesis mainly derives from studies at the neuromuscular junction, but new work is beginning to reveal analogous mechanisms at CNS synapses. Recent data suggest that nascent nerve terminals contain clusters of some, but not all, presynaptic proteins, and upon contact with a dendrite they induce the clustering of early postsynaptic proteins at the site of contact. Through a process of mutual cross talk, the pre- and postsynaptic sides coordinately orchestrate the maturation of the entire synapse, including spine development.(26)
Molecular Composition of Spines
As in all cells, cytoskeletal proteins take the lead in determining the shape and stability of spines. The spine cytoskeleton is mainly based on actin filaments (F-actin), since intermediate filaments and microtubules are absent from nearly all spines. Spines are unusually rich in F-actin compared to other parts of the neuron. Pharmacological manipulations that inhibit F-actin dynamics alter spine shape and motility.(10),(27) and experimentally-induced spine collapse can be prevented by stabilizing actin filaments.(23)
A growing roster of F-actin binding and regulatory proteins have been found in spines (Figure 2,3), and many of these have been shown to regulate spine shape.(2),(3)
Figure 2. Some of the Major Components of Dendritic Spines.
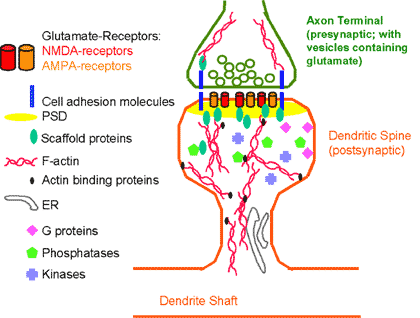
Note that the relative abundance of various molecules differs greatly from that shown in this schematic diagram, and many components are omitted for simplicity.
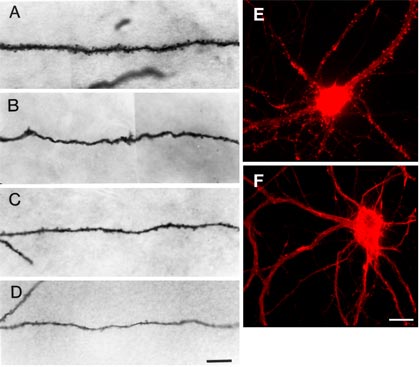
Epilepsy model. Comparison of spine numbers on dendrite segments of hippocampal pyramidal cells from a control and three experimental rats. A shows a dendritic segment from a control rat. Note the high density of spines along the dendrite. B-D are examples of dendritic segments of three pyramidal cells from three separate epileptic rats. In B, spine density is severely reduced. In C and D, spines are present but greatly reduced in number. Often the diameters of dendritic shafts are also reduced in experimental rats, as seen in D. Scale bar, 10μm. (Reprinted from Journal of Neuroscience Vol. 18, p8362: Jiang M, Lee CL, Smith KL, and Swann JW, Spine loss and other persistent alterations of hippocampal pyramidal cell dendrites in a model of early-onset epilepsy," with permission from The Journal of Neuroscience, copyright 1998 the Society for Neuroscience.)
E-F. Effect of the glutamate agonist NMDA on dendritic spines of neurons cultured from embryonic rat hippocampus. Both cells were fixed and stained with fluorescently-tagged phalloidin, which selectivley binds to filamentous actin (F-actin). E. Control neuron, illustrating the high concentration of F-actin in dendritic spines, seen here as small protrusions and F-actin-rich foci along the dendrites. F. Neuron incubated for 5 min with 50μm NMDA. Note that F-actin is selectively lost at spiny protrusions, but retained (even somewhat increased) in dendrite shafts. This loss of F-actin in spines is accompanied by collapse of the spine structure. Scale bar, 20μm.
Examples include Rho family GTPases(28) and proteins that regulate them, such as SPAR (spine-associated GTPase activating protein for Rap)(29) and kalirin (a guanine nucleotide exchange factor for Rac);(30) the scaffolding protein shank;(31) and the actin binding proteins drebrin;(32) cortactin,(33) and profilin.(34)
Changing the expression levels of these proteins has been shown to alter spine numbers, shape, or motility. It is likely that many such proteins will eventually be linked to diseases that affect synaptic function. Indeed, it was recently shown that the gene encoding LIMK-1, an actin regulatory molecule, is one of the approximately 20 genes deleted in patients with Williams syndrome, a developmental disorder characterized by cardiovascular and cognitive deficits.(35) LIMK-1 is a protein kinase that responds to Rac by phosphorylating and thereby inactivating the actin-depoylmerizing factor ADF/cofilin.(36) LIMK-1 knockout mice and Williams syndrome patients exhibit abnormal dendritic spines.(37)
The heads of dendritic spines contain a specialized organelle called the postsynaptic density (PSD), which sits directly across the cleft from the presynaptic active zone, where synaptic vesicles are clustered (Figure 2). The PSD is a supramolecular assembly of glutamate receptors, scaffolding molecules and enzymes.(33) A significant component of the PSD is the calcium/calmodulin-dependent protein kinase type II (CaMKII), which may play both structural and enzymatic roles within the PSD.(2) Scaffolding molecules, such as PSD-95, spinophilin, and Homer are proteins that have no enzymatic function of their own but which serve to concentrate receptors and their downstream effector molecules into the correct assembly for efficient signal transduction.(38) This protein meshwork may also contribute to regulation and maintenance of spine structure.
Despite their small size, dendritic spines also contain molecular machinery for many other functions, including local control of membrane trafficking, protein synthesis, post-translational processing, and protein degradation. Most dendritic spines in the cerebellum and about 30% of spines in the hippocampus (those with the largest head volumes) contain endoplasmic reticulum (ER) in the spine neck, which exists either as loose tubular structures or as an organelle called the spine apparatus that consists of stacks of membrane cisternae.(39)
ER is thought to contribute not only to trafficking of membrane proteins but also to regulation of calcium dynamics within the spines. Among other functions, ER releases calcium in response to calcium influx into the spines and thereby amplifies the calcium signal.(8),(40) In addition, many dendrites have polyribosomes just beneath the spine neck, and there is growing evidence that protein synthesis and protein degradation can occur locally within spines.(41),(42),(43) Disruption of local protein synthesis has emerged as a potential mechanism for cognitive deficits in Fragile X syndrome, because Fragile X mRNA is targeted to dendrites, and the Fragile X mental retardation protein itself regulates protein synthesis.(44)
Function of Spines
Spines clearly contain specialized collections of receptors and signaling complexes, and they are uniquely associated with excitatory CNS synapses. However, the function of spines is not completely understood. Indeed, many types of synapses, both excitatory and inhibitory, do not occur on spiny protrusions, so clearly a spine is not a pre-requisite for a functional synapse. It is likely that the existence of the unique spine structure confers some special property to the synapse that would be lost if the spine structure collapsed. Early work addressed this issue using computer modeling and suggested that the electrical properties of synapses (for example, the passive spread of synaptic current) are different for a synapse occurring on a spine as compared to one occurring on a dendrite shaft.(45) Such models predicted that changes in spine shape would modulate current spread, leading some investigators to speculate that changes in spine shape might underlie learning and memory and other forms of synaptic plasticity, such as long-term potentiation (LTP). Later studies, however, questioned whether such changes would attain sufficient magnitude to significantly impact synaptic strength [reviewed in (5)].
A current, widely accepted idea is that spines act as tiny, individual biochemical compartments. By virtue of its narrow neck, a spine retains second messengers and other small, diffusible molecules that are generated at the site of synaptic contact. Such "biochemical compartmentalization" has been directly demonstrated for small diffusible dyes and also for the important ions sodium and calcium. (46),(47),(48),(49) For calcium in particular, local control of concentration is crucial in orchestrating a cell's physiological state.
Biochemical compartmentalization by spines provides two advantages. First, it allows small molecules to attain high concentrations in the post-synapse prior to their diffusing away. Second, compartmentalization biochemically and therefore functionally isolates individual synapses from the parent dendrite.(5) This latter concept means that molecular signals generated at one spine are restricted to that synapse, and are not able to influence a neighboring synapse on the same dendrite. This property of dendritic spines probably contributes crucially to the phenomenon of "input specificity," whereby one set of axonal inputs to a neuron can induce temporary or long-lasting effects in the synapses that are specific to that set of post-synaptic contacts, and do not induce the same changes at other synapses driven by different axons impinging upon the same neuron.(50)
The function of the spine neck in limiting calcium diffusion from spine head to adjacent dendrite has also been postulated to play a protective role in excitotoxic injury during, for example, seizures or ischemic conditions.(51) In such diseases, the excitatory neurotransmitter glutamate is released in excess, leading to overly strong activation of glutamate receptors and injurious calcium overload in the postsynaptic neurons. Such calcium overload is a major factor in excitotoxic neuronal cell death.(52),(53) The diffusion barrier of the spine neck should limit the rise of calcium concentration in dendrite shaft and cell body, and thereby possibly protect from cell death. Indeed, studies using a neuronal cell culture model support an excitoprotective role for spines (S. Graber and S. Halpain, unpublished). Since spines are found only on excitatory synapses, such a neuroprotective role could have been an additional factor driving their evolutionary development.
Spine Abnormalities in Neurological Disease
A wide variety of human neurological and psychiatric diseases exhibit loss of spines or abnormalities in spine shape, paralleled by impaired cognitive function. Spine loss or distortion is associated with stroke, epilepsy, trauma, dementia, brain tumors, schizophrenia, major depression, substance abuse, and normal aging.(5),(54),(55),(56) Brain regions showing spine loss typically correlate with brain systems affected in the disease state. For example, schizophrenic patients exhibit decreased spine density selectively in prefrontal cortical pyramidal neurons.(57) Spines are also significantly reduced in some animal models of chronic depression, an effect reversed by antidepressants.(58)
A direct link between spine loss and specific pathological events is best established for acute disorders such as seizures and stroke, which involve excitotoxic injury to the brain. Human epileptic patients display spine loss and other dendritic abnormalities.(55) This observation is supported by work in animal models.(59) Tetrodotoxin induction of epileptic seizures in rats reduces spine density on hippocampal pyramidal cells to 20-35% of normal controls.(60) Pilocarpine- or bicuculline-induced seizures also cause spine loss.(61) Presumably, the excessive activation of glutamate receptors during seizures or post-ischemia reperfusion probably swamps the calcium-buffering capacity of the spines, causing spine retraction or collapse.(62)
Excitotoxic injury to spines can be studied in cultured neurons by addition of glutamate analogs such as NMDA to the culture medium.(23) In such models, spine loss appears very rapidly, within 5-10 min of the onset of the stimulus. Spine loss can also be induced in culture by addition of a calcium ionophore, revealing a crucial role for glutamate-induced calcium influx in triggering spine collapse. However, such excessive stimulation of glutamate receptors and calcium also can induce neuronal cell death hours or days later. Thus, spine loss could be merely an early morphological step in the progression toward neuronal death. However, our laboratory recently showed that intense but sub-lethal activation of glutamate receptors in cultured neurons can induce widespread spine loss in the absence of cell death (S. Graber and S. Halpain, unpublished). This dissociation between spine loss and cell death is consistent with the idea that spine loss itself might contribute to abnormal brain function after injury, even when the neurons themselves are preserved.
Chronic neurodegenerative diseases such as Alzheimer's and Parkinson's often exhibit dendritic changes, including spine loss, as an early step in disease progression. Emerging evidence suggests that initial changes in motor and cognitive function in such diseases are due to subtle alterations in synaptic function, rather than reflecting neuronal loss per se. For example, it is now widely believed that the initial stages of Alzheimer's disease actually reflect disrupted synaptic function that greatly precedes the later decline in neuronal numbers.(63) Dendrites and spines also degenerate prior to cell loss in some murine cerebellar ataxias.(64),(65) Spine loss occurs without neuronal death in a mouse model of Huntington's Disease,(66) although the human disease clearly shows cell loss at later stages. It was previously thought that, as in neurodegenerative diseases, normal aging also involved a decline in neuronal cell numbers; however, more recent studies suggest that this is not the case, and instead that synapse numbers are reduced.(67) Even if spine loss per se is not the underlying cause of cognitive decline in either aging or neurodegenerative disease, it is likely that changes in dendritic spines contribute to impaired cognition.
Perhaps the most compelling link between spine abnormalities and cognitive dysfunction occurs in mental retardation, where brain structure and neurochemistry are grossly normal, implying that synaptic function is somehow impaired via more subtle defects.(68) Patients with non-specific mental retardation, Down's syndrome, and Patau's syndrome have fewer spines and spines with thin, elongated necks and enlarged heads.(54),(68),(69),(70),(71),(72),(73),(74) Fragile X syndrome patients have abnormal neurites, thin, elongated spines, and smaller synaptic contacts than normal,(75),(76),(77) and a mouse model of the disease, in which the gene encoding Fragile X mental retardation protein (FMRP) is deleted, similarly exhibits abnormal spine shapes.(78) Moreover, a LIMK-1 knock-out mouse, a model of Williams syndrome, a disease that results from a micro-deletion in chromosome 7 that affects this and other genes, also exhibits spine abnormalities.(37)
Before the intrinsic motility of spines was appreciated (see above), it was thought that in these developmental diseases spines fail to make the transition from filopodia to spines.(8) In light of more recent observations on spine motility and morphing in adult neurons, it is possible that at least some of these diseases are caused by dysregulation of spine motility or shape maintenance, rather than initial spine production.
Furthermore, it is probably naive to assume that cognitive decline will always correlate with reduced spine number, since researchers still do not understand the relationship between numbers of spines and overall mental performance. In this regard, it is interesting to note that postnatal neurological changes following chronic placental insufficiency were reported to include increased spine density.(79)
Together the above observations demonstrate a strong correlation between abnormal cognition and impairments in the production, elimination, or maintenance of spines. However, they do not prove that the spine abnormalities directly lead to deficits, rather than being secondary to some other event. Indeed, since it is likely that neural activity throughout life regulates both the number and shape of spines, it remains possible that spine abnormalities are the effect, rather than the cause, of cognitive deficits. Finally, much work is required to establish the precise relationship between the shape and physiological properties of dendritic spine synapses.
Molecular Mechanisms In Spine Loss: Potential Use in Fighting Disease
Elucidation of the enzymes or other molecules that participate in spine loss may provide therapeutic targets that help to prevent spine loss or enhance spine recovery. The main cytoskeletal elements of spines are actin filaments. F-actin disassembly appears necessary for spine loss, since drugs that stabilize F-actin prevent glutamate-induced spine collapse.(23) A novel pathway for inducing spine loss in mature neurons was recently described.(80) Neuronal activity was shown to activate a protein kinase called serum-inducible kinase (SNK), which in turn phosphorylates the actin regulatory protein SPAR. Phosphorylated SPAR becomes subject to proteolysis through the ubiquitin-mediated proteasome pathway. This elegant mechanism represents the first model that links extracellular signals to specific protein degradation in the context of dendritic spine regulation.
At present it is unclear whether the molecular pathways in excitotoxic spine loss are distinct from those regulating physiological spine turnover. Prevention of excessive glutamate receptor activity would be one means to prevent excitotoxic spine loss, but glutamate antagonists have complex and deleterious side effects.(81) A new generation of compounds is under development that may selectively reduce hyper-stimulation of glutamate receptors without interfering with normal levels of activity.(81) Such compounds may hold promise for therapies in many types of neuronal injury, including spine loss.(82)
Promotion of spine recovery may also be a meaningful goal in the therapeutic setting. In our cell culture model, we observe that spines can recover from sub-lethal stimuli, and that blockade of glutamate receptors dramatically promotes spine recovery, even when added after spine collapse has occurred (Graber and Halpain, unpublished). It is likely that in this case the synaptic connections are not broken, and that spines reemerge at their original sites of contact with the pre-synaptic terminal. In other words, network connectivity may be preserved during the process of spine loss and recovery under certain circumstances. In light of the possible neuroprotective effect of spines, preservation or recovery of spines in both chronic and acute neurological diseases seems a reasonable therapeutic goal.
Summary and Outlook
Dendritic spines are key players in information processing in the brain. Changes of spine shape and wholesale spine turnover provide mechanisms for modifying existing synaptic connections and altering neuronal connectivity. While neuronal cell death in acute and chronic neurodegenerative diseases is clearly an important factor in cognitive or motor function decline, in this Cyberounds® we make a case that the loss of dendritic spines, in the absence of cell death, may also play an important role in the loss of cognitive and motor function in disease and aging.
Because spines may function in neuroprotection as well as cognition, it may be a clinically worthwhile goal to prevent spine loss and/or enhance spine recovery. Progress in defining the molecular pathways in spine loss and in providing conditions favorable for spine recovery may lead to therapeutic advances in treating neurodegenerative disease and acute neurological damage. Finally, advances in understanding spine development and stabilization may yield insights into mental retardation and psychiatric disorders.