Course Authors
Brian Meldrum, M.B.B. Chir., Ph.D.
Release Date: 04/27/2004
Upon completion of this Cyberounds®, you should be able to:
Describe the roles of glutamate ionotropic receptors in epileptic discharges and the changes in glutamate receptor function in acquired epilepsy in animals and man
Describe the anticonvulsant effects in animal models of AMPA and NMDA receptor antagonists
Discuss the effects of selective glutamate metabotropic agents on seizure phenomena.
The prevalence of epilepsy is about 1 in 200 (4-8 /1,000) in industrialized countries. The incidence is highest in the first year of life and in old age. The prevalence appears to have remained relatively constant in the last two to four decades, perhaps because improvements related to the declining role of bacterial and parasitic infections are offset by an aging population. The personal and social burden of epilepsy is substantial. Currently available drug treatment fails to suppress seizures in about one-third of patients.
Pathophysiology
The clinical syndromes grouped under the title "epilepsy" continue to be defined by the excessively synchronous discharge of groups of neurons.(1) This pathophysiology takes two principal forms which present clinically as absence seizures (generalized, non-convulsive) or as convulsive seizures (focal or generalized). In absence seizures, the synchronization (at 3 Hz in man, 6-8 Hz in rodents) involves oscillation within thalamocortical circuits with the EEG spike-and-wave activity corresponding to isolated spikes on a background of hyperpolarization in cortical pyramidal neurons.(2) The characteristic pathophysiology observed with intracellular recording in focal or generalized convulsive epilepsy is a "paroxysmal depolarizing shift," a giant excitatory postsynaptic potential with a burst of action potentials riding on it (see Figure 1).
Figure 1. Intracellular Record of a "Paroxysmal Depolarizing Shift" Induced by Electrical Stimulation in a Pyramidal Neuron in a Rat Hippocampal Slice.
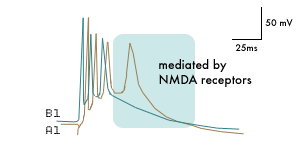
A1. Response induced by both AMPA and NMDA receptor activation.
B1. The early components produced by AMPA receptor activation remain but the later components due to NMDA receptor activation are abolished.
Adapted from Dingledine, R., et al., (1986). NMDA Receptors and Interictal Bursting, pp. 179.
The initial component of the giant excitatory potential characteristic of convulsive seizures results from an inward flux of Na+ through AMPA receptor channels, while the second component of the convulsive seizure is provoked by Ca++ and Na+ entering through NMDA receptor channels, as shown by the use of AMPA and NMDA receptor selective antagonists.(4) The pharmacology of absence seizures(5) is, accordingly, different from that of convulsive seizures. Exacerbation of absence attacks can be induced by some antiepileptic drugs potentiating gamma-amino butyric acid (GABA) (e.g., tiagabine and vigabatrin) and by some drugs prolonging inactivation of Na+ channels (e.g., phenytoin and carbamazepine). Some drugs (ethosuximide, trimethadione) selectively treat absence seizures. Some drugs (valproate, lamotrigine) are broad-spectrum and can treat complex partial seizures and absence attacks.
Genetics
The last decade has seen marked advances in our understanding of the genetic and developmental causes of epilepsy. Single gene defects involving ion channels have been identified in some rare familial syndromes, including benign familial neonatal convulsions (K+ channel, genes KCNQ(2),(3)) and a syndrome, GEFS+, in which febrile seizures precede other forms of generalized epilepsy (Na+ channel, α1, α2, β1 subunits, genes SCN1A, SCN2A, SCN1B).(6)(7)
Mutations involving the GABAA receptor subunits have also been identified. Mutations in the γ2 subunit have been described in families with febrile convulsions and other generalized seizures(8)(9) and in the Α1 subunit in a family with juvenile myoclonic epilepsy.(10) This syndrome, characterized by myoclonic episodes on awakening in adolescents, accounts for about 8% of cases of epilepsy. Its genetic basis is evidently complex.
Absence epilepsy is familial but its genetic basis in man is unknown. An allelic association of juvenile absence epilepsy with a GluR5 kainate receptor gene polymorphism has been reported, but as yet there is no identified mutation in a glutamate ionotropic receptor gene that gives rise to a generalized epilepsy syndrome. In contrast, specific gene defects have been identified in several inherited mouse models of absence seizures. These mouse disorders are predominantly recessive, with genetic defects in subunits of the voltage sensitive calcium channels, e.g., tottering α1 subunit; lethargic β4 subunit; stargazer γ2 subunit; ducky α2δ2 subunit.(2)
Disorders of neuronal migration associated with focal or general abnormalities in cortical lamination are now recognized as an etiological factor in a significant proportion of cases of focal and generalized epilepsy that were earlier considered to be "idiopathic". Several specific gene defects have been identified in the more severe forms, such as a mutation in doublecortin (a novel 40 kDa predicted protein) that is responsible for Double cortex/X-linked lissencephaly (smooth brain).(11)
This Cyberounds® will focus primarily on the convulsive syndromes because of the clear evidence of the critical involvement of glutamatergic activity in these syndromes.(12)
The Role of Glutamate Receptors
Glutamate metabotropic receptors play an important role in epilepsy(13) through the direct excitatory effect of Group I receptor activation and the presynaptic action of Group II and III receptors to decrease glutamate release.(14) There have been two main approaches to evaluating the role of glutamate receptors in epilepsy. One concerns changes in the expression or function of receptors or their subunits in animal or human syndromes of epilepsy. The other concerns the use of pharmacological agents acting selectively on specific glutamate (ionotropic or metabotropic) receptors in models of epilepsy. In spite of impressive preclinical data, the pharmacological approach has yet to make a major impact on therapeutic practice.
Receptor Changes
Our most detailed knowledge of changes in glutamate receptor function comes from two sources:
- kindled seizures in rodents , a model of acquired epilepsy in which repeated electrical stimulation initially produces only brief afterdischarges eventually produces full seizures, and a permanently enhanced seizure sus susceptibility and
- tissue slices prepared from anterior temporal lobectomy specimens removed from patients with drug refractory temporal lobe epilepsy. These patients commonly show hippocampal or Ammon's Horn Sclerosis (AHS), with loss of neurons in the CA1 and CA3 zones and the endfolium.(15)
They commonly give a history of a prolonged or complex febrile convulsion in infancy or early childhood or other potential precipitating event.(15)(16) The molecular biological data from kindled rats and patients with AHS are surprisingly similar, reinforcing the concept that kindled seizures provide a model for understanding acquired limbic seizures in man (see Figure 2).
Figure 2. Diagram of a Glutamatergic Synapse.
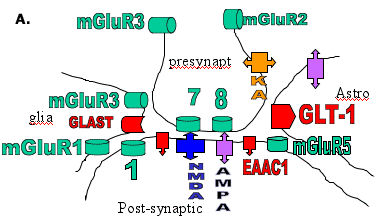
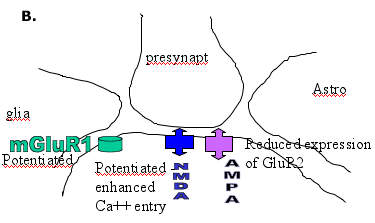
A. the predominant location of glutamate receptors and transporters
B. the major receptor changes contributing to enhanced glutamate excitatory responses.
Changes in NMDA receptor responses have been repeatedly described in amygdala-kindled rats. Enhanced Ca++ entry or Ca++ conductance changes are seen in dentate granule cells and in CA3 pyramidal neurons.(17) Increases in mean open times, burst lengths and cluster duration are seen in isolated granule cells.(18) The precise molecular basis of these changes is not known but may involve altered phosphorylation of the NMDA receptor. There is an increase in the NR2B mRNA in the supraoptic nucleus and limbic regions of amygdala-kindled rats (at one month post-kindling).(19) The pharmacology of NMDA receptors is clearly altered in the kindled rat, as shown by changes in the binding properties of competitive NMDA antagonists.(20) In vitro studies suggest that the potency of competitive NMDA antagonists is diminished(21), but, in vivo, the motor side effects of NMDA antagonists are enhanced in kindled rats.(22)
Kindled animals also show changes in the expression and function of AMPA receptors. There is a reduction in the expression of GluR2 subunits in the piriform cortex(23) and a reduction in the contribution of AMPA receptors containing GluR2 to AMPA-induced currents.(24) A reduction in GluR2 mRNA and protein expression are also seen in association with enhanced seizure susceptibility following lithium/pilocarpine induced status epilepticus.(25) In human temporal lobe epilepsy, there is an alteration in the expression of GluR2 in dendrites in the hippocampal CA3 region that appears to be associated with epileptogenicity.(26) There is also evidence that autoantibodies to an AMPA receptor subunit (GluR3) play a role in the pathogenesis of some severe progressive forms of epilepsy. The autoantibodies can be detected in the plasma in children with Rasmussen's syndrome(27) and also children with "catastrophic" epilepsy.(28)
Mutations involving glutamate receptors have yet to be identified in familial epilepsies in man. The possibility of such a mechanism is, however, clearly established by studies in mice with an editing deficiency in GluR2(GluRB) such that calcium entry is enhanced when hippocampal AMPA receptors are activated.(29) Such mice show an early onset epilepsy syndrome that proves lethal. This clearly indicates that some deficiency in the mechanism by which RNA-edited GluR2 subunits reduce the Ca++ entry induced by AMPA receptor activation could be a factor contributing to acute epileptogenesis.
The clearest evidence for changes associated with epileptogenesis is provided by kindled rats in which enhanced expression and function of group I metabotropic receptors are seen. Transient changes in both mGlu1 and mGlu5 mRNA are seen in the hippocampus(30) but a more lasting change in mGluR1 mRNA occurs in the supraoptic nucleus.(31) Functional enhancement of Group I responses is observed in biochemical and electrophysiological studies. Thus phospho-inositide hydrolysis induction by quisqualate or other Group I mGlu receptor agonists is enhanced in the amygdala(32),(33) and the depolarizing effect of Group I agonists is potentiated.(34),(35) The mechanism of potentiation is not clear, but in the case of mGluR1 may result from increased expression of Homer1a (an "activity-inducible short-splice variant" of the Homer protein) decreasing internalization of the receptor by longer forms of Homer (a postsynaptic density (PSD) protein).(36) It may simply result from increased expression of mGluR1 (mRNA and protein) as reported in the dentate gyrus in temporal lobectomy specimens.(37)
In rats, antisense oligonucleotides that reduce the expression of mGluR1 decrease the rate of kindling, which suggests an involvement of mGluR1 in epileptogenesis.(38) Studies in gene knockout mice support a role for mGluR7 in seizures and in the antiepileptic effect of Group III antagonists.(39) There is electrophysiological evidence for a loss of function of Group III receptors, both in kindled rats(40) and in temporal lobe epilepsy patients with hippocampal (Ammon's horn) sclerosis.(41)
NMDA and AMPA Antagonists as Anticonvulsants
NMDA antagonists have long been recognized as potent anticonvulsants in animal models of epilepsy, especially sound-induced seizures in DBA/2 mice and GEPrats or photically-induced seizures in baboons.(42),(43) They are less effective against fully-kindled seizures(44),(45) The competitive antagonist, D-CPPene, in a preliminary add-on trial for drug-resistant complex partial seizures, failed to produce clinical improvement but was associated with more severe neurological side effects.(46)
AMPA competitive and non-competitive antagonists (2,3-benzodiazepine negative allosteric modulators) are also effective in animal models of epilepsy, including reflex seizures, maximal electroshock and kindled seizures.(47),(48) Problems relating to pharmacokinetics and toxicity have hampered the clinical development of competitive antagonists. A non-competitive antagonist, the 2,3-benzodiazepine, talampanel, has been in clinical trial for epilepsy.(49)
Kainate receptors have their highest expression within the hippocampus. Kainate, or domoate, given systemically or intra-cerebroventricularly induce limbic seizures. The availability of compounds with selective actions at kainate receptors and their subtypes has recently improved.(50) A compound that selectively antagonizes KA ionotropic channels containing GluR5 (LY 382884) fails to block maximal electroshock seizures but potently blocks limbic seizures induced by 6 Hz corneal stimulation.(51)
Current Drugs and Glutamate Receptors
Although no pure glutamate receptor antagonists have been introduced clinically, it is clear that several antiepileptic drugs have actions on glutamate receptors that may contribute to their antiepileptic effect. Thus felbamate decreases the excitatory effect of glutamate at NMDA receptors(52) with some evidence for selectivity at the NR2B subunit.(53) Actions on AMPA/KA receptors may contribute to the effects of phenobarbitone and topiramate.(54) Chronic administration of valproate leads to changes in AMPA receptor subunit expression in the hippocampus.(55) Effects of valproate on NMDA receptor mediated effects are also described.(56)
Glutamate Agents: Animal Models of Epilepsy
Compounds that act selectively at Group I receptors as agonists are convulsant.(57),(58),(59) These include (S)-3,5-dihydroxyphenylglycine which acts equipotently on mGlu1 and mGlu5 and (RS)-2-chloro-5-hydroxyphenylglycine which acts selectively on mGlu5.(60) Compounds that are Group I antagonists are anticonvulsant in a variety of animal models, including absence epilepsy models.(61) This is seen with competitive antagonists acting preferentially on mGlu1, but also with allosteric antagonists acting on mGlu5.(62),(63),(64)
Group II agonists such as LY 354740 and LY 379268 are anticonvulsant in many epilepsy models including absence models.(65) Although classical Group III selective agonists sometimes produce proconvulsant effects, novel Group III agonists with some subtype selectivity, such as phosphonophenylglycine and (S)-3,4-dicarboxyphenylglycine, are effective anticonvulsants in reflex epilepsy in rodents.(66),(67),(68) No effects of anti-epileptic drugs on metabotropic receptors have been described, but the possibility that they act at an allosteric site on metabotropic receptors has not been adequately tested.
Glutamate Receptors and Epileptic Brain Damage
Prolonged seizures in children and adults lead to a characteristic pattern of cell loss in the hippocampus involving neurons in the CA1 and CA3 zones(15) and pyramidal neurons in lamina 2/3 of the cortex.(69),(70) This pathology arises from an excessive influx of Na+ and Ca++ following prolonged activation of AMPA and NMDA receptors.(71),(72) It can be prevented by NMDA receptor antagonists even when they do not alter the total duration of seizure activity.(73),(74) The cascade of events following the glutamate receptor activation and calcium overload is similar to that occurring in ischemic brain injury.(75) and leads to both necrotic and apoptotic cell death. The pattern of selective vulnerability of neurons in the forebrain and hippocampus is probably at least partially attributable to the high expression of NMDA receptors containing NR2A and NR2B subunits.
Glucocorticoids can potentiate excitotoxicity by NMDA receptor activation(76) and apoptosis (cell death) induced in hippocampal neurons by glucocorticoids can be prevented by NMDA receptor antagonists and by mGluR antagonists.(77) Antiepileptic drugs with a neuroprotective action might decrease the progressive neuronal loss and cognitive decline observed in some patients with epilepsy.(78),(79) Felbamate with its action on NMDA receptors and topiramate acting on AMPA/KA receptors would appear to be candidates, but clinical trials to establish such an effect present severe difficulties.
Menstrual Cycle and Seizures
Female gonadal and adrenal steroid hormones act on neuronal receptors to modify seizure phenomena.(80) Some endogenous metabolites of progesterone, 3α-hydroxy-5α/β-pregnane steroids, "GABA-steroids,", act at a site on the GABAA receptor to potentiate the effect of GABA on chloride conductance.(81) Acutely, this effect diminishes convulsive seizures but potentiates absence attacks. Chronically, GABA steroids show tolerance and withdrawal effects, and catamenial (monthly) epilepsy is related to a reduced efficacy of GABA associated with withdrawal of the GABA steroids in the menstrual phase.(81) In contrast, estradiol is proconvulsant in experimental models of epilepsy. Potentiation of NMDA-receptor-mediated excitatory currents by estradiol, demonstrated in hippocampal slices,(82),(83) is likely to contribute to this effect.
Prospects for Future "Glutamatergic" Drugs
Our improved understanding of (a) the genetic basis of a small number of familial syndromes and (b) changes in the expression and function of neurotransmitter receptor molecules has yet to yield therapeutic dividends. It has, nevertheless, reinforced the concept that ion channels and neurotransmitter receptors are appropriate targets for novel antiepileptic drugs, and, most importantly, made us aware that we should target the altered molecules associated with the epileptic state. Thus it has probably been a major error to have used (over the last 65 years) normal animals with unmodified channels and receptors for screening purposes. Future screening programs need to employ in vitro expression systems for the altered molecules found in animals or patients with epilepsy and in vivo animals with genetic or acquired epilepsy syndromes (as opposed to normal animals with electrically or chemically-induced seizures).
We also need to understand the molecular basis of drug resistance in epilepsy. Is it related to increased expression of drug transporter molecules(84) or is it caused bydue to loss of effect of particular drugs at their molecular sites of action?(85) Why do some patients develop drug resistance during the course of their illness,(86) while other patients with adult onset are drug resistant from the beginning of their illness?,(87),(88)
Prospects for glutamate ionotropic receptor antagonists remain uncertain, but it is still possible that an NMDA antagonist, either with a low affinity for the open channel or with subtype selectivity, could avoid the problems of toxicity seen with potent competitive or high-affinity open channel blockers.(89) Selective AMPA antagonists are still under investigation as potential anti-epileptic drugs (AEDs). They may have clinical utility either alone or in combination with other drugs acting on glutamatergic synapses.
Preclinical studies suggest a possible role for Group I metabotropic antagonists or Group II and III metabotropic agonists as antiepileptic drugs, but problems related to long-term toxicity remain to be addressed. It is, however, clear that a combination of effects, either at different sites within the glutamatergic synapse or at an additional non-glutamatergic site, can be effective. In preclinical models, an enhanced therapeutic effect can be obtained by combining NMDA and AMPA antagonism or combining effects on ionotropic and metabotropic receptors.(90) The evidence that an action on glutamate ionotropic receptors may contribute to the effect of some current AEDs is given above. The next decade is likely to see a greater emphasis on the role of glutamatergic mechanisms both in epileptogenesis and in the therapy of epilepsy.