Course Authors
Josef Anrather, V.M.D., and Costantino Iadecola, M.D.
Release Date: 03/09/2004
Upon completion of this Cyberounds®, you should be able to:
Learn about the neurotoxicity associated with NMDA receptors and related mechanisms
Learn about the role of NMDA-induced neurotoxicity in ischemic brain injury
Understand the therapeutic potential of NMDA receptor inhibitors in ischemic stroke.
Ischemic brain injury results from the concerted action of multiple pathogenic factors that act sequentially after the onset of ischemia (Figure 1).(1),(2) The ischemic cascade is triggered by a sudden and severe reduction in cerebral blood flow. Ischemic neurons release large amounts of the neurotransmitter glutamate that activates glutamate receptors, especially the N-methyl-D-aspartate (NMDA) type.
Figure 1. The Mechanisms of Ischemic Brain Injury.
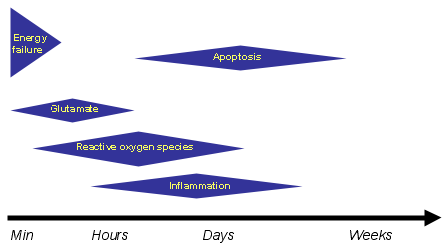
The mechanisms of ischemic brain injury are multiple and act sequentially at different time points after induction of ischemia. Activation of glutamate receptors is an early event in the ischemic cascade that is critical for the expression of ischemic injury.
Stimulation of NMDA receptors leads to an increase in intracellular calcium, which, in turn, activates signaling pathways responsible for the short- and long-term cellular and molecular events leading to neuronal cell death (apoptosis). These include, for example, production of reactive oxygen species, inflammation and apoptosis. Thus NMDA receptor activation is thought to be a crucial step in the ischemic cascade.
The purpose of this Cyberounds® is to provide a brief overview of the role of glutamate receptors in ischemic brain injury and of their therapeutic potential.
The Concept of Excitotoxicity
Nearly five decades ago, Lucas and Newhouse(3) described the phenomenon glutamate-induced neuronal cell death. They found that systemic injection of L-glutamate into developing mice caused the destruction of the neuronal retinal layer. These studies were later confirmed and expanded by Olney(4) who coined the term "excitotoxicity" to define the neurotoxicity deriving from excessive neuronal excitation by glutamate.
Subsequent studies demonstrated that excitotoxicity is involved in ischemic brain injury. Cerebral ischemia was found to release toxic amounts of glutamate in the extracellular space,(5),(6) and inhibition of the NMDA receptor attenuated brain injury in animal models of ischemic stroke and in models of hypoxic-ischemic injury in neuronal cultures(7),(8) [see ref. (9) for a review].
Amelioration of ischemic injury has also been reported by blocking NMDA receptors using antisense oligonucleotides designed to downregulate the expression of a critical subunit of the receptor(10) and by a blocker of glutamate release.(11) More recently, excitotoxicity has been found to participate in other brain pathologies, including trauma, seizure, multiple sclerosis, and neurodegenerative diseases (amyotrophic lateral sclerosis, Alzheimer's, Huntington's and Parkinson's disease).(12),(13),(14),(15)
NMDA Receptor Subtypes and Excitotoxicity
The NMDA receptor is a ligand gated ion channel requiring two agonists (glycine and glutamate) to bind simultaneously for channel opening. It is a tetrameric protein complex composed of two NR1 and two NR2 subunits [see ref. (16) for a review]. There are several NMDA receptor subunits (NR1, NR2A through D and NR3).
Both NR1 and all NR2 subunits are membrane proteins with three transmembrane regions and an intramembrane loop between the first and second transmembrane domain. The extracellular N-terminal domain contains ligand-binding sites and is multiply glycosylated. The intracellular C-terminus is responsible for anchoring the receptor to specific cellular sites and for its interaction with diverse signaling modules.(17)
The NR1 subunit has a short cytoplasmic tail and is several hundred amino acids shorter than members of the NR2 family of proteins. In addition, there are four C-terminal (NR1-1 to NR1-4) and one N-terminal NR1 splice variants that result in eight different proteins. This process produces a very heterogeneous receptor population that explains in part the differences in susceptibility of distinct neuronal populations to excitotoxic damage.
For example, cortical neurons expressing mainly NR1/NR2A and NR1/NR2B receptors are more susceptible to glutamatergic cell death than cerebellar neurons, which have high levels of NR1/NR2C composed channels. Accordingly, transfection of non-neuronal cells with NR1 and NR2A or NR2B receptor subunits was able to induce L-glutamate-triggered cell death. In contrast, cells transfected with NR1/NR2C or NR1/NR2D receptors were much less susceptible to excitotoxic cell death, even though whole cell currents in these cells were similar in magnitude to those in cells expressing NR1/NR2A or NR1/NR2B receptors.(18)
These studies define the NR2A and NR2B subunits as the main transducer of excitotoxicity. However, the NR1 subunit is also required. The NR1 subunit is an important site for modulation of NMDA receptor signaling by the serine protease tissue plasminogen activator (tPA). The data suggest that tPA cleaves the NR1 subunit near the N-terminus, a modification that potentiates calcium flux through the receptor and enhances cytotoxicity both in vitro and in vivo.(19) This finding has important clinical implications because tPA is routinely administered to patients with ischemic stroke in an attempt to dissolve the arterial blood clot causing the stroke.
Cellular Calcium and Glutamate Excitotoxicity
Cellular calcium has been linked to a variety of potentially damaging signaling pathways (Figures 2, 3), including activation of calcium-dependent endonucleases that provokes DNA degradation, breakdown of cytoskeleton structure by calcium sensitive proteases (e.g., calpain), production of arachidonic acids and subsequent free radical production, nitric oxide (NO) generation by NO synthase (NOS) and mitochondrial damage by calcium overload. Ca2+ activates NOS by facilitating the binding of the signaling protein calmodulin to the enzyme, a step that is required for its catalytic activity.
Figure 2. Signaling Pathways Linked to NMDA Receptors.
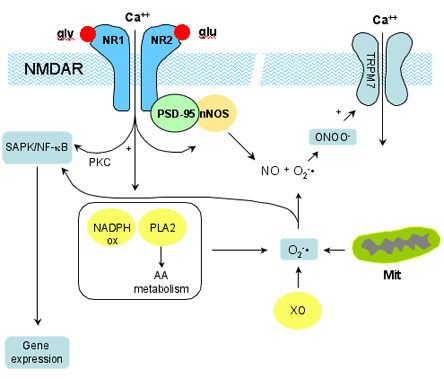
Binding of glutamate (glu) and glycine (gly) to the NMDA receptor allows Ca2+ entry into the neuron. Ca2+ activates nNOS, which is associated with NR2 through PSD-95, and leads to NO production. Activation of other Ca2+ dependent enzymes, such as NADPH oxidase and phospholipase A2 (PLA2), leads to production of superoxide (O2-). PLA2 metabolizes membrane phospholipids into arachidonic acid (AA). AA is further metabolized by lipoxygenases, cyclooxygeneases, and p450 epoxygenases. O2-, which is also produced by xanthine oxidase (XO) and mitochondria (Mit), reacts with NO to form peroxynitrite (ONOO-). Peroxynitrite has many biological effects (see Figure 3) including activation of the TRPM7 channel, which produces Ca2+ influx and contributes to the late phase of excitotoxicity. Ca2+ influx induces gene expression, possibly, via Ca2+-dependent protein kinase C, leading to activation of stress activated protein kinases (SAPK), and of the transcription factor NK-kB, pathways that can also be activated by reactive oxygen species.
Although other ions such as sodium and chloride have been linked to excitotoxic neurodegeneration in certain models,(20),(21) Choi and colleagues(22),(23) described the dependence of glutamate neurotoxicity on Ca2+ influx. They showed that, while the initial cell swelling upon transient glutamate exposure is dependent on the presence of extracellular Na+, most neurons degenerate over the next 24 hours even in the absence of Na+. However, if Ca2+ was omitted during the glutamate challenge, a large proportion of neurons were protected.
These observations led to the "calcium-mediated excitotoxicity" hypothesis. Several studies showed a high degree of correlation between the amount of cellular Ca2+ load and glutamate toxicity.(24),(25),(26) However, this correlation seems to be only valid for glutamate induced Ca2+ uptake. Other routes of Ca2+ influx such as activation of voltage sensitive calcium channels (VSCC) by high extracellular potassium concentrations are not neurotoxic even if they produce a Ca2+ uptake that is comparable or exceeds that resulting from NMDA receptor stimulation.(25),(26) While glutamate produces a linear correlation between Ca2+ and neurodegeneration, NMDA elicited Ca2+ accumulation is not linearly related to cell death. Thus, at low NMDA concentrations (<30 μM), NMDA is able to induce Ca2+ influx, but fails to produce neurodegeneration.(25)
The NMDA receptor, as other receptors, is embedded in a highly specialized membrane subdomain called the post-synaptic density (PSD), and its subunits contact several proteins of this structure.(27) One of these proteins is PSD-95/SAP90 (Post Synaptic Density-95/Synapse Associated Protein 90) and it has a crucial role in mediating NMDA receptor triggered neuronal cell death. Disruption of PSD-95/NMDA receptor interactions, either by using a blocking peptide or by reducing PSD-95 expression with antisense oligonucleotides, attenuates glutamate dependent neuronal cell death in vitro and reduces ischemic brain damage in vivo.(28),(29) This approach does not block Ca2+ influx.
In the context of excitotoxicity, PSD-95 works as an adapter that links neuronal NOS (nNOS), the enzyme that synthesizes NO, to the NMDA receptor.(29) Thus, the high Ca2+/Calmodulin concentrations reached in close proximity of the NMDA receptor after channel opening can activate nNOS. Increased NO is then believed to react with superoxide (O2-) to form peroxynitrite (ONOO-), which is cytotoxic.
However, blocking NMDA and other glutamate receptors might not be sufficient to counter cell death. In a model of OGD (Oxygen Glucose Deprivation), glutamate receptor blockage was able to rescue neurons from 1-hr but not from 2-hr OGD.(30) Prolonged OGD induced Ca2+ influx that was not inhibited by NMDA and AMPA antagonists or by L-type Ca2+ channel blockers. Rather, TRPM7, a member of the transient receptor potential (TRP) cation channel family, has been shown to underlie the late stage Ca2+ influx in prolonged OGD. Ca2+ flux through this channel depends on reactive oxygen species (ROS) and NO, suggesting that ONOO- is involved in its regulation.(30)
Silencing the gene that encodes for TRPM7 reduced late Ca2+ influx and cytotoxicity caused by OGD.(30) Therefore, two mechanistically distinct Ca2+ influx pathways, both leading to neurotoxicity, can be identified in the neuronal injury produced by OGD. While the "early" Ca2+ influx is triggered by the NMDA receptor, the late influx is linked to the TRPM7 channel.
Reactive Oxygen Species (ROS) and Effectors of Excitotoxicity
As discussed above, ROS might be instrumental for NMDA receptor mediated cytotoxicity. It was first shown by J. Bockaert's group that NMDA enhances O2- production in neurons, which is positively correlated to neuronal cell death.(31) Several enzymatic pathways can generate ROS following NMDA receptor activation. Membrane bound NADPH oxidase,(32) cyclooxygenases and lipooxygenases,(33) xanthine oxidase,(34) and mitochondrial respiratory chain(35),(36) have been implicated as sources for NMDA receptor induced radicals (Figure 2).
However, even though ROS are a central component of glutamate-induced neurotoxicity, source and makeup of radical species may differ in neuronal subpopulations. For example, in cerebellar granule cells the toxicity resulting from glutamate receptor activation depends on ROS but not on NO,(31),(33) while in cortical and hippocampal neurons cell death requires both ROS and NO.(30),(37)
How do ROS modulate or execute neuronal death? While O2- is the main species produced by excitotoxic stimuli, O2- itself is not very reactive, but it can form highly toxic adducts by reacting with hydrogen peroxide in a transition metal-catalyzed reaction to yield hydroxyl radical and by reacting with NO to produce ONOO-. The latter is especially important since several studies found nNOS inhibition to be protective from excitotoxicity.(29),(38),(39) ONOO- is highly reactive promoting protein dysfunction by adding NO2 to cyclic aminoacids, a process termed nitration.(40)
Figure 3. Main Effector Pathways Leading to Cell Death Following Activation of NMDA Receptors.
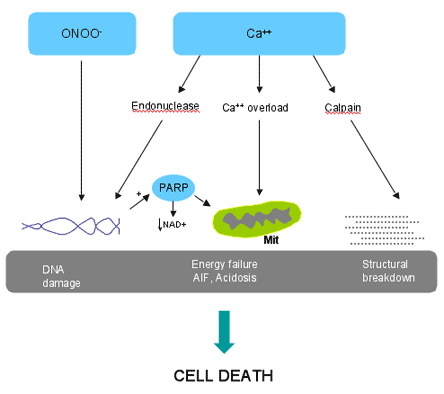
Endonucleases, activated by Ca2+, and peroxynitrite (ONOO-) produce DNA damage, which, in turn, activates the DNA repair enzyme PARP. PARP is neurotoxic by consuming the essential substrate NAD+, and by inducing the release of apoptosis inducing factor (AIF) from mitochondria (Mit). Ca2+ accumulation in mitochondria results in energy failure and formation of excess H+ ions (acidosis). Ca2+-dependent activation of proteases, such as calpain, causes damage to structural proteins (tubulin, spectrin, focal adhesion kinases, etc.), which contribute to the breakdown of the cell.
One of the targets of ONOO- is TRPM7, the channel responsible for the late Ca2+ influx in OGD-induced excitotoxicity (see above), which is presumably opened by nitration.(30) Furthermore, ONOO- induces DNA strand breaks which, in turn, activate Poly(ADP-ribose) polymerase-1 PARP-1(41) PARP-1 is part of a family of DNA binding enzymes that transfer up to 200 molecules of ADP-ribose to a variety of nuclear proteins and is involved in many cellular functions such as DNA repair and replication, gene transcription, cell proliferation and cell death [reviewed by ref. (42)].
With respect to neuronal survival, PARP inhibition has been shown to be beneficial in several in vivo and in vitro models of excitotoxicity(43) (reviewed by ref. (41)). Beside the potential energy depletion that is triggered by NAD+ consumption,(42) PARP also has more specific effects on regulating excitotoxic cell death by liberating apoptosis-inducing factor (AIF) from mitochondria, which initiates a form of neuronal cell death that is independent of caspases, cysteine proteases that mediate programmed cell death(44) (Figure 3). In addition to being involved in executing the cell death program initiated by excitotoxic signals, ROS might also sustain the excitotoxic process by increasing glutamate release.(45) Thus, ROS are multi functionally involved in excitotoxic cell death.
Effects on Gene Transcription
NMDA receptors activate protein kinase pathways that lead to gene transcription. While cyclic AMP (cAMP)-dependent protein kinase (PKA) activates the transcription factor CREB (cAMP-response element-binding protein), which results in transcription of neuroprotective genes [reviewed in ref. (46)], activation of stress activated protein kinases (SAPK), including members of the JNK family (c-jun N-terminal kinase), and activation of the transcription factor NF-kB are thought to be noxious by up-regulating such genes as COX-2 and p53.
JNK3 seems to be the major effector within this gene family since JNK3 deficient mice are protected in models of cerebral ischemia and glutamate-induced neuronal cell death.(47),(48) Additionally, inhibition of JNK with substrate-competing peptides, markedly reduced lesion volumes after transient cerebral ischemia.(49) NF-kB, while protective and anti-apoptotic in other cell systems, is thought to induce expression of gene products that are promoting cell death in neurons.(50) For example, NF-kB dependent up-regulation of p53 and subsequently of the cell death protein Bax, has been implicated as an effector mechanism in glutamate-mediated neuronal cell death.(51),(52),(53)
NMDA receptor-induced expression of COX-2 might increase ROS production on one hand, and generate prostaglandins (PGs) on the other. PGE2, the major COX-2 metabolite,(54) has been reported either to be protective or to exacerbate neurotoxicity (e.g., ref. (55),(56)). These conflicting data probably reflect the fact that PGE2 acts on four separate classes of receptors (EP1-4), which have very different signaling profiles.(57) Thus, activation of the EP2 receptor seems to promote neuronal survival via cAMP signaling,(58) while activation of EP1 receptors seems to be toxic, presumably, by exacerbating calcium excitotoxicity(56) (Anrather and Iadecola, unpublished observations).
Peri-infarct Depolarizations
Glutamate receptor activation also participates in the mechanisms of cerebral ischemia by inducing peri-infarct depolarizations (PID). Following occlusion of the middle cerebral artery, the brain tissue undergoes recurrent waves of depolarization and repolarization resembling the phenomenon of cortical spreading depression (CSD) of Leao.(59),(60),(61) CSD is induced by focal cortical injury, such as a needle stab or topical application of KCl, and is characterized by a wave of depolarization that spreads over the cerebral cortex at a rate of 2-3 mm/min.(62) The depolarization is associated with a profound increase in blood supply and energy consumption that is needed to reestablish the ionic gradient following the depolarization.(63),(64)
PID differs from CSD in that it is not associated with increases in cerebral blood flow.(65) PID can be attributed to excessive glutamate release following ischemia,(59) and is blocked by NMDA receptor antagonists.(61),(66) PID enhances the energy failure by subjecting the ischemic brain to the additional metabolic load needed to reestablish the ionic gradients.(65),(67),(68) Therefore, PID-induced energy failure is another mechanism by which activation of glutamate receptors contributes to ischemic brain injury. The recent finding that PID occurs in the human brain after stroke or trauma underlies the clinical relevance of this phenomenon.(69)
Non-NMDA Receptors
Although most studies have focused on NMDA receptors, there is evidence that non-NMDA receptors also play a role in the pathogenesis of ischemic damage. For example, the toxicity mediated by AMPA receptors may affect predominantly cell types enriched in these receptors, such as oligodendrocytes.(70) Because of this, AMPA receptor antagonists, unlike NMDA antagonists, protect cerebral white matter from ischemia,(71),(72) a feature with important therapeutic implications. The role of metabotropic glutamate receptors has not been extensively characterized and both protective and destructive effects have been reported.(73),(74),(75),(76) Overall, non-NMDA receptor antagonists are potentially useful and warrant further preclinical and clinical evaluation.
Clinical Trial Failures
Despite the powerful neuroprotection exerted by NMDA and non-NMDA receptor antagonists in models of cerebral ischemic injury, clinical trials using these agents in stroke patients have not reproduced the beneficial effects reported in animal models. The reasons for this discrepancy have been debated extensively in the literature.(1),(77),(78),(79) The emerging view is that the lack of correlation between the outcome of clinical trials and animal experiments can be attributed to several factors related to the specific drug used, to the design of the clinical trials and to the animal models used in preclinical experiments.
First, in the case of glutamate receptor antagonists, the concentrations of the inhibitors that conferred protection in animal models could not be achieved in humans because of psychogenic side effects of the drugs. Therefore, the inhibitors could not be given to stroke patients at an effective concentration.
Second, in these clinical trials, the inhibitors were administered many hours after stroke onset, while the toxic effects of glutamate occur in the early stages of cerebral ischemia (Figure 1). Therefore, the inhibitors were administered at a time when the damage was no longer caused by activation of glutamate receptors but by independent downstream events, such as free radical damage, inflammation or apoptosis (Figure 1).
Third, most models of cerebral ischemia use young healthy animals, typically rodents, in which neuroprotection may be relatively easy to achieve. Human stroke occurs in individuals with many confounding factors, including advanced age, diabetes, atherosclerosis, hypertension, that worsen ischemic injury. Thus, the protection offered by glutamate receptor antagonists is likely to be less efficacious in elderly humans with pre-existing diseases. Further complicating the issue is the fact that neuroprotection is more easily attained in rodents that in higher order mammals.(80)
Fourth, in animal models the neuroprotection exerted by a certain agent can be precisely assessed by directly measuring the volume of brain injury. In contrast, clinical trials typically measure outcome by using neurological scales based on clinical examination. Although neurological examination is of paramount importance in the management of stroke patients, it is not as sensitive a measure of outcome as the volume of injured brain.
Therefore, there are many reasons for the lack of concordance between the results of clinical trials with glutamate receptor antagonists and those of animal experiments. This discrepancy can be reconciled by the development of inhibitors lacking neuropsychological side effects, by improving the design of clinical trials to include shorter therapeutic windows and more sensitive outcome measures, and by developing animal models that reflect better the clinical reality of stroke patients.
Conclusions
Activation of glutamate receptors, especially the NMDA type, plays a central role in the mechanisms of ischemic brain injury. Most of these effects are mediated by influx of Ca2+ that triggers a wide variety of signaling mechanisms that, either directly or though changes in gene expression, lead to neuronal death. Thus, interventions to down-regulate glutamate receptors and their downstream signaling pathways hold great promise in the treatment of cerebral ischemia. Because glutamate is a neurotransmitter that is essential for the function of the normal brain, indiscriminate inhibition of glutamate receptors will impair crucial aspects of brain function, such as learning and memory, and limit their clinical use.
As noted in the previous section, the neurological side effects of NMDA receptor antagonists have been a major problem in clinical trials of neuroprotection. Therefore, development of more selective inhibitors that target predominantly the deleterious aspects of glutamate signaling would be highly desirable. In addition, targeting downstream signaling pathways involved in neurotoxicity would have great therapeutic potential. Thus, ROS scavengers, NOS inhibitors, COX-2 inhibitors and approaches to block selected transcriptional events initiated by glutamate receptor activation, such as JNK, offer new pathways to neuroprotection.
In many stroke patients, the major neurological deficits derive from interruption of white matter tracts. Therefore, targeting white matter ischemia specifically with AMPA receptor inhibitors would enhance the effectiveness of neuroprotective strategies. Finally, considering the mutifactorial nature of cerebral ischemic injury (Figure 1), the need to combine multiple therapeutic approaches is becoming increasingly evident.(81),(82) Therefore, glutamate receptor antagonists could be used in combination with reperfusion therapy using tPA, or with pharmacological agents targeting other pathogenic mechanisms.(81),(82) Although the feasibility and effectiveness of these novel approaches need to be validated in stroke patients, there are reasons to be cautiously optimistic about the future of neuroprotection in the treatment of this devastating brain disease.