Course Authors
Bassem A. Bejjani, M.D., Richard Alan Lewis, M.D., M.S.
Release Date: 11/02/1998
Upon completion of this Cyberounds®, you should be able to:
Discuss the molecular basis of the different forms of glaucoma
Describe the genetic and clinical heterogeneity of the glaucomas
Counsel patients with glaucoma and their relatives.
Background
The glaucomas are a heterogeneous group of insidious visual disorders that combine elevated intraocular pressure (IOP) and optic nerve damage that can lead to blindness.(3) As a group, they are the second leading cause of blindness (after cataract) in the world, affecting an estimated 70 million individuals.(25) In the United States, 2.5 million individuals are estimated to have open-angle glaucoma, half of whom are unaware of their disease. The disorder is present in 2% of those over 40, and its prevalence increases with age. African-Americans have a four-fold increase risk over Caucasians,(26) suggesting the genetic background affects the expression of the disease. The hereditary predisposition to glaucoma was first suspected in the mid-1800's following the description of glaucoma in two sisters.(7)
Clinically, the glaucomas are classified by the etiology (primary or secondary), by the anatomy of the anterior chamber (open angle or closed angle) and by the age of onset (infantile, juvenile, or adult). The most common forms, primary open-angle glaucoma (POAG), also known as chronic open-angle glaucoma (COAG), have onset in mid-adulthood and relentless slow progression that culminates in blindness, unless appropriate medical and/or surgical therapies are instituted. The juvenile and infantile glaucomas are Mendelian disorders that are both severe clinically and difficult to manage.
Autosomal dominant juvenile-onset open-angle glaucoma (JOAG) (MIM # 137750) has a typical onset in the second or third decade, high IOP, poor responsiveness to medical therapy and frequent requirement for filtering surgery to control intraocular pressure and the attendant optic neuropathy.
Primary Congenital Glaucoma (PCG) (MIM # 231300) is a clinical and genetic entity clearly distinct from the juvenile forms.(5),(16) Autosomal recessive PCG occurs when the developmental anomalies of the angle prevent adequate drainage of aqueous humor, so that the IOP is elevated.(9) The coats of the infantile eye are sufficiently plastic that they stretch with this elevated pressure and yield an enlarged globe (buphthalmos).
In the present Cyberounds®, the genetics of the various forms of glaucoma will be discussed and the molecular bases of PCG and JOAG will be reviewed in detail. The emerging knowledge of the molecular mechanisms leading to these two pediatric conditions may be instrumental in understanding the pathogenesis of the more common, adult-onset POAG that affects 2.5 million individuals in the United States. Additionally, an understanding of the molecular bases of these conditions is invaluable for accurate genetic counseling of the patients and their families.
Primary congenital glaucoma (PCG)
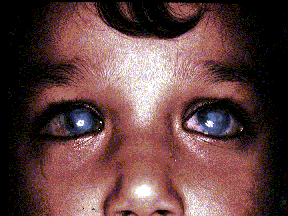
PCG is a devastating autosomal recessive disorder that manifests in the neonatal or infantile period and is caused by unknown developmental defect(s) of the anterior chamber angle.(11),(15) Although PCG is the most common form of glaucoma in infancy, nothing secure is known about its specific embryologic pathogenesis, despite studies of various animal models.(8),(9),(10),(12),(14) However, there is consensus about its classic clinical characteristics.(11) These include: elevated IOP; enlargement of the globe, particularly the anterior segment; secondary edema and opacification of the cornea with rupture of Descemet's membrane; thinning of the anterior sclera and atrophy of the iris; anomalously deep anterior chamber; structurally normal posterior segment except for sequential optic atrophy; and photophobia, blepharospasm, and hyperlacrimation. Both eyes are involved in about half of the cases.
The earlier the defect appears, the less favorable the prognosis. Depending on when treatment is instituted, there may be reduced visual acuity and/or restricted visual fields. Therapy is primarily surgical with a variable success rate, as more than one surgical intervention may be necessary to control intraocular pressure in a number of these patients. Thus, significant morbidity is associated with both this condition and the currently available treatment options. In untreated cases, blindness occurs invariably (Figure 1).
Infantile glaucoma, however, may also be a feature of a number of well recognized syndromes (e.g., aniridia, anterior segment dysgenesis, microcornea, Nance-Horan syndrome, Lowe syndrome, Congenital Hereditary Endothelial Dystrophy, Neurofibromatosis Type 1, and Sturge-Weber syndrome) and segmental aneusomy syndromes. We will exclude all these conditions from our discussion and will limit our review to the classical isolated, autosomal recessive form of Primary Congenital Glaucoma.
PCG has an estimated incidence of 1:10,000 in the United States. It is much more common in the Middle East (1:2,500)(2) and in the Gypsy population of Slovakia (1:1250).(24) PCG is genetically heterogeneous with at least three loci identified. Genetic linkage studies have mapped a PCG locus (GLC3A) on the short arm of chromosome 2 (2p21),(28) and another locus (GLC3B) on the short arm of chromosome 1 (1p36).(1) A number of families did not show any evidence of linkage of the disease gene to either GLC3A or GLC3B. Thus, at least a third locus remains to be mapped. We will review the currently available information on each of these loci.
GLC3A
Initial linkage of a locus for PCG to markers on 2p21 was shown in 1995 in a group of eleven Turkish and Canadian families. Six families did not show any linkage to 2p21.(28) Later, three different mutations in CYP1B1, the gene for cytochrome P4501B1, were described in five Turkish families whose disease was linked previously to the 2p21 locus.(31) Subsequent surveys confirmed that 2p21 is a major locus for PCG in Saudi Arabian(6) and Slovakian families.(24) Three distinct point mutations in CYP1B1 were defined as the cause for PCG in 24 of 25 Saudi Arabian families studied.
Analysis of the haplotype and mutational data from these families revealed that 19 individuals with two mutant alleles did not show any evidence for disease at the time of examination, suggesting reduced penetrance for selected alleles in this population.(6) Further analysis of the mutations in the Saudi population revealed eight distinct mutations that seem to correlate with tribal ancestry and to have arisen on different genetic backgrounds (haplotypes) [manuscript in preparation].
By contrast, mutational analysis in the Gypsy Slovakian population showed genetic homogeneity for a unique and novel CYP1B1 mutation and suggested that a single mutation in a founder is responsible for all PCG in that population (V. Ferak, personal communication).
These studies suggest that mutations in CYP1B1 account for 85-90% of all PCG cases in various ethnic groups and across vast geographic areas.
All PCG-causing mutations reported to date are predicted either to truncate the protein and result in null alleles or to alter significantly the putative enzymatic function of the protein by substituting highly conserved residues with established functional importance to both structure and function of the protein.
CYP1B1 is a dioxin-inducible member of the cytochrome P450 gene superfamily(34),(35),(36) with a yet undefined role in ocular development. CYP1B1 and other drug-metabolizing enzymes seem to regulate the steady state of ligands that affect growth and differentiation.(20),(21) Mutations that alter the function of CYP1B1 presumably disrupt that steady state and result in either abnormally high levels of these ligands or prolonged exposure of the developing tissues to these postulated effectors of growth and development. Either event could potentially cause structural aberrations in the developing tissues.Future studies of CYP1B1 and the identification of its putative ligands will contribute to the understanding of the pathogenesis of PCG and to the elucidation of important pathways in human ocular development. Such knowledge should provide pharmacologic targets for the antenatal modulation or postnatal therapy of PCG and possibly other forms of glaucoma.
GLC3B
GLC3B was mapped to 1p36 in 4 of 8 families that did not show linkage to GLC3A.(1) The remaining four families did not show linkage to markers on 1p36, suggesting that there is at least one more, yet unmapped, locus for PCG. No candidate gene has been identified.Juvenile-onset open-angle glaucoma (JOAG)
Juvenile-onset open-angle glaucoma (JOAG) is a rare autosomal dominant form of glaucoma. The identification of a gene responsible for JOAG might shed some light on the multifactorial pathogenesis of the more common, but genetically more complex POAG.
A locus for JOAG was first mapped to 1q21-1q31 in a large American pedigree.(30) This finding was later confirmed in a number of families from various ethnic and geographic backgrounds.(17),(27),(38),(13),(19) These mapping efforts culminated in the identification of GLC1A.(33) This gene, myocillin [also called TIGR (Trabecular meshwork Inducible Glucocorticoid Response)], was cloned initially by differential library screening with selection criteria based on the induction pattern of a new protein found in human trabecular meshwork (the loosely packed cells that act as a drain for the aqueous as it exits the eye through the canal of Schlemm ) cultures after prolonged exposure to glucocorticoids.(23)
The role of myocillin in normal and glaucomatous eyes is unknown. It is postulated that myocillin may regulate aqueous flow in the anterior chamber and that mutations in this gene upset this steady-state, result in aqueous accumulation and lead to increased intraocular pressure and glaucoma.(22) Deleterious mutations in myocillin have been also detected in 4-5% of a sample of patients with POAG with no known family history of the disease.(4)
An interesting finding was recently reported in a large consanguineous Canadian family segregating myocillin mutations and autosomal dominant POAG.(18) The authors noted that patients with the homozygous myocillin mutation K423E did not develop glaucoma, whereas their heterozygous parents and siblings expressed the disorder. This phenomenon is best explained by postulating that K423E has a dominant negative effect on protein function and that normal molecular physiology is restored in the homozygous individuals by virtue of intragenic complementation.
While testing for myocillin mutations in adult-onset glaucoma is not advocated, it could be a worthwhile endeavor in families with an unequivocal autosomal dominant transmission of the condition or with a severe, juvenile-onset form of the disease. The search for genes that could play a role in optic nerve excavation and atrophy (the direct cause of blindness in glaucoma) is likely to provide additional insights into the pathogenesis of this condition.
Additional "glaucoma genes"
To date, additional loci for "glaucoma genes" have been mapped to various chromosome regions. These are summarized as Table 1.
| Locus | Chromosomal location | Phenotype | Reference |
| GLC1B | 2cen-q13 | Moderate increase in IOP | (Stoilova et al., 1996)(32) |
| GLC1C | 3q21-q24 | Moderate increase in IOP | (Wirtz et al., 1997)(39) |
| GLC1D | 8q23 | Mild increase in IOP | (Trifan et al., 1998)(37) |
| GLC1E | 10p15-p14 | Normal tension glaucoma | (Sarfarazi et al., 1998)(29) |
It is possible that each of these loci contains one or more gene(s) that could play a role in the pathogenesis of POAG. The hunt for these genes continues and the dissection of the molecular bases of the glaucomas promises to be one of the most interesting stories of this decade.